
HPLC法测定诺氟沙星胶囊的有关物质
2022-10-11 08:28李丽云黎艳刚朱海云南省药物研究所云南省中药和民族药新药创制企业重点实验室昆明650111
江西中医药 2022年9期
★李丽云 黎艳刚 朱海(云南省药物研究所/云南省中药和民族药新药创制企业重点实验室 昆明 650111)
诺氟沙星又称氟哌酸,是人工合成的第三代喹诺酮类抗菌药,1979年日本杏林制药公司发现了该化合物并申请了专利。1983年该药由默沙东作为申请人在意大利最初上市生产,当时采用片剂形式,商品名为Noroxin。1984年由杏林制药作为申请人在日本上市生产,所用商品名为Baccidal,同样采用片剂形式,该药目前仍然在日本上市。诺氟沙星胶囊于1987年在国内上市,目前有批文690余条[1],已有两家制药企业通过仿制药质量和疗效一致性评价。诺氟沙星在抗菌方面具备广谱性,特别在抵抗需氧GNB(革兰阴性杆菌)方面具备高效性,于体外可有效抑制肠杆菌科细菌中的多数菌属,同时体外可抵御多种耐药菌,对因敏感菌引发的尿路感染、伤寒、淋病、肠道感染、前列腺炎、其它沙门菌感染等具有治疗作用[2]。
本文通过对中国药典[3]所收载的诺氟沙星胶囊有关物质检测方法及欧洲药典所收载、参考文献[4]所记载的诺氟沙星有关物质检测方法进行了考察和对比,发现上述方法存在杂质控制不严、溶剂峰对主峰及杂质峰的测定产生干扰、梯度洗脱过程中容易出现基线波动等不足。因此,此项研究经色谱条件的筛选,完成诺氟沙星胶囊相关物质测定方法的建立,同时对其开展方法学验证。
1 仪器与试剂
Agilent 1260高效液相色谱仪,奥豪斯EX125ZH电子天平,梅特勒 DELTA 320型pH计。
诺氟沙星对照品及杂质A对照品、环丙沙星(Ciprofloxacin)对照品、依诺沙星(Enoxacin)对照品,均购于中国食品药品检定研究院;诺氟沙星杂质D对照品及杂质2对照品均购于TLC公司;诺氟沙星杂质H对照品、杂质B对照品、杂质K对照品及杂质E对照品均购于CATO公司;诺氟沙星胶囊(自研)。甲醇为色谱纯,水为纯化水;其它试剂均为分析纯。
2 方法与结果
2.1 色谱条件
采用安捷伦 ZORBAX SB-C18色谱柱(5 μm× 4.6 mm×250 mm);流动相A为0.015 mol/L磷酸溶液(由三乙胺将pH调节为3.0±0.1)-甲醇(85∶15),流动相B为甲醇,根据表1实施线性梯度洗脱处理;柱温、流速分别设定为30 ℃、1.0 mL/min;测定波长为262 nm与278 nm。
2.2 溶液的配制
稀释剂:即“2.1”项下的流动相A。
供试品溶液:取适量诺氟沙星胶囊内容物(与15 mg诺氟沙星相当),精密称定,移至100 mL量瓶内,用2 mL盐酸溶液(0.1 mol/L)溶解,再通过流动相A定容至100 mL,摇匀后滤过,即得供试品溶液。
各杂质对照品贮备液:分别取诺氟沙星杂质D、K、B、E 、2、A、H对照品约15 mg,精密称定,移至100 mL量瓶内,用2 mL盐酸溶液(0.1 mol/L)溶解,再通过流动相A定容至100 mL,摇匀,即得对照品贮备液。
峰定位溶液:取诺氟沙星对照品约15 mg,精密称定,移至100 mL量瓶内,用2 mL盐酸溶液(0.1 mol/L)溶解,分别量取上述杂质对照品贮备液各4 mL,再通过流动相A定容至100 mL,摇匀,作为峰定位溶液。
系统适用性溶液:分别取环丙沙星对照品和依诺沙星对照品约15 mg,精密称定,移至50 mL量瓶内,用2 mL盐酸溶液(0.1 mol/L)溶解,加流动相A定容至50 mL,摇匀;再取诺氟沙星对照品约15 mg,精密称定,并移至100 mL量瓶内,用2 mL盐酸溶液(0.1 mol/L)溶解,再加入1 mL上述混合液,之后通过流动相A定容至100 mL,摇匀,即得系统适用性溶液。
空白辅料溶液:取适量诺氟沙星空白辅料(与15 mg的诺氟沙星相当),精密称定,再移至100 mL量瓶内,通过2 mL盐酸溶液(0.1 mol/L)溶解,再通过流动相A定容至100 mL,摇匀后滤过,即得空白辅料溶液。
2.3 系统适用性试验
取“2.2”项下制备的峰定位溶液、系统适用性溶液分别进样,对色谱图进行记录。所得结果见图1,系统适用性溶液和峰定位溶液中各杂质与主峰、杂质与杂质之间分离完全,分离度均大于2.0,理论塔板数均大于4 000,符合系统适用性要求。
2.4 专属性试验
2.4.1 辅料干扰试验 取“2.2”项下制备的峰定位溶液、供试品溶液、空白辅料溶液与稀释剂分别进样,对色谱图进行记录。所得结果见图1,各杂质峰处均未见空白辅料与稀释剂出峰,表明辅料与溶剂皆不会影响对诺氟沙星胶囊的杂质测定。
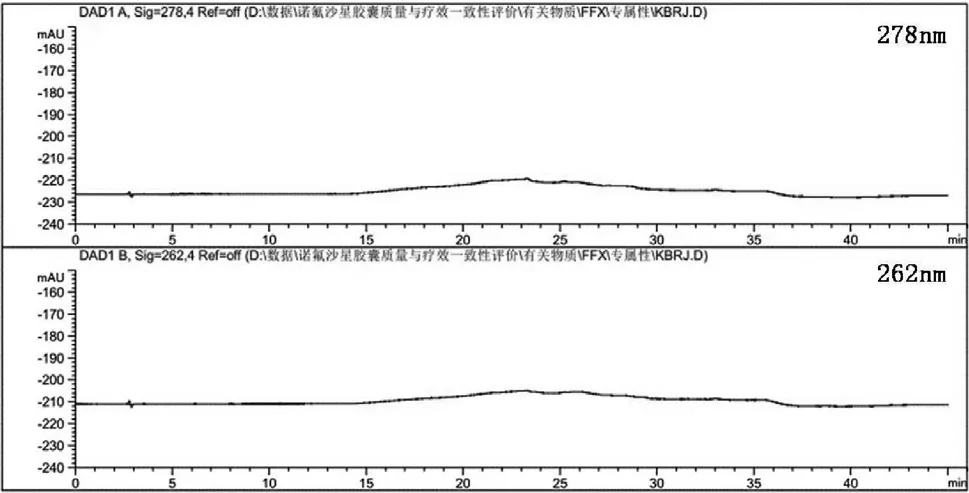

图1 典型色谱图
2.4.2 强制降解试验 取“2.2”项下供试品溶液和空白辅料溶液,分别置于2 mol/L盐酸溶液中、105 ℃条件下酸破坏4 h;2 mol/L氢氧化钠(NaOH)溶液内,105 ℃温度时碱破坏4 h;30%双氧水溶液0.1 mL中,105 ℃条件下氧化破坏3 h;105 ℃高温下破坏24 h;光照(5 000±500)lx条件下1个月,处理完成后分别进样,记录色谱图。结果表明诺氟沙星胶囊在酸、碱、氧化、高温、光照条件下均相对稳定,产生的降解产物较少,各降解产物峰与诺氟沙星主峰能完全分离,峰纯度合格,满足专属性强制破坏要求。
2.5 线性和范围
取诺氟沙星对照品约15 mg,精密称定,移至100 mL量瓶内,用2 mL盐酸溶液(0.1 mol/L)溶解,再通过流动相A定容至100 mL,摇匀,即得诺氟沙星对照品贮备液;分别精密量取“2.2”项下各杂质对照品贮备液1 mL以及上述诺氟沙星对照品贮备液1 mL,移至25 mL量瓶内,通过流动相A定容至25 mL,摇匀,即得对照品混合溶液;对此混合液分别进行不同量的准确量取:0.01、0.015、0.02、0.03、0.04、0.06、0.1 mL,逐一移至液相色谱仪,对色谱图进行记录。横坐标(X)、纵坐标(Y)分别为“对照品的进样量(单位:ng)”“峰面积”,开展线性回归,得到回归方程与相关系数r。见表2。
2.6 定量限和检测限
分别量取“2.2”项下各杂质对照品贮备液及“2.5”项下诺氟沙星对照品贮备液适量,用流动相A稀释至信噪比为10∶1和3∶1的浓度,精密量取上述对照品溶液各0.02 mL,注入液相色谱仪,记录色谱图。见表2。

表2 诺氟沙星及已知杂质的线性关系及灵敏度测定结果
2.7 精密度试验
2.7.1 仪器精密度 精密量取1 mL“2.5”项下诺氟沙星对照品贮备液,移至25 mL量瓶内,用流动相A定容至25 mL,摇匀,即得对照品溶液。精密量取上述对照品溶液0.02 mL,注入液相色谱仪,共进行6次不间断进样,记录色谱图,通过主峰峰面积与保留时间的RSD波动,考察仪器于此色谱条件下的精密状况。结果显示,关于对照品溶液,其主峰峰面积、保留时间RSD值依次是1.88%、0.69%,可见仪器于此色谱条件下具备良好精密度表现。
2.7.2 重复性 依照“2.2”项下制备供试品溶液的方法,完成6份供试品溶液的制备,之后分别准确吸量0.5 mL,移至100 mL量瓶内,通过流动相A定容至100 mL,摇匀,即得对照溶液;分别准确吸量此对照溶液与供试品溶液0.02 mL,移入液相色谱仪,对色谱图进行记录,根据外标法,通过峰面积对已知杂质含量展开求解,得到杂质K的RSD值是1.97%,可见此法具备良好重复性。
2.7.3 中间精密度 依照“2.2”项下制备供试品溶液的方法,由不同操作者,借助不同仪器于不同时间,取12份批次相同的样品进行重复检测,考察方法的中间精密度,其中第1天的数据引用重复性项下的结果,结果显示杂质K的RSD值等于3.93%,可见此法具备良好中间精密度。
2.8 准确度试验
分别量取“2.2”项下各杂质对照品贮备液及“2.5”项下诺氟沙星对照品贮备液2 mL,移至50 mL量瓶内,通过流动相A定容至50 mL,摇匀,即得对照品混合贮备液;之后量取2.5 mL,移至50 mL量瓶内,通过流动相A定容至50 mL,摇匀,即得对照品混合溶液。取空白辅料适量,精密称定,置50 mL量瓶中,分别加入对照品混合贮备液2.0、2.5和3.0 mL,通过流动相A定容至50 mL,摇匀后滤过,取续滤液制备不同浓度供试品溶液,低、中、高浓度依次为80%、100%、120%,各浓度皆重复配样3份,分别移入液相色谱仪,对色谱图进行记录,借助外标法,经由峰面积对已知杂质含量展开计算。结果显示,诺氟沙星杂质D、K、B、E、2、A、H及诺氟沙星的平均回收率为99.19%、107.95%、98.85%、103.43%、106.16%、106.05%、102.35%、99.04%,RSD值为0.60%、2.40%、1.28%、1.51%、1.28%、1.29%、0.98%、2.64%,表明本法检查有关物质的准确度良好。
2.9 溶液稳定性试验
依照“2.2”项下制备供试品溶液的方法进行供试品溶液的制备,静置于室温中,分别于0、4、8、12、16、20、24 h精密吸量上述样品溶液0.02 mL,分别注入液相色谱仪,记录色谱图。结果显示,供试品溶液内总杂质、最大单杂、已知杂质皆未见明显改变,同时无新杂质形成,可见供试品溶液于室温静置24 h依然具备稳定性。
2.10 耐用性试验
经由调整流速、流动相比例、磷酸盐酸碱度、柱温、厂家不同、批号不同的色谱柱来考察拟定色谱条件的耐用性。准确吸量0.02 mL供试品溶液,移至液相色谱仪,对色谱图进行记录。结果表明当流速变动±10%、柱温变动±5 ℃、磷酸盐pH值变动±0.2、流动相的比例变动±2%、改变色谱柱时,对测定结果不造成影响,说明本法对流速、柱温、磷酸盐pH值、流动相的比例、色谱柱的耐用性良好。
2.11 样品测定
按照经方法学验证的有关物质检查方法,对自研诺氟沙星胶囊的有关物质进行检查。见表3。
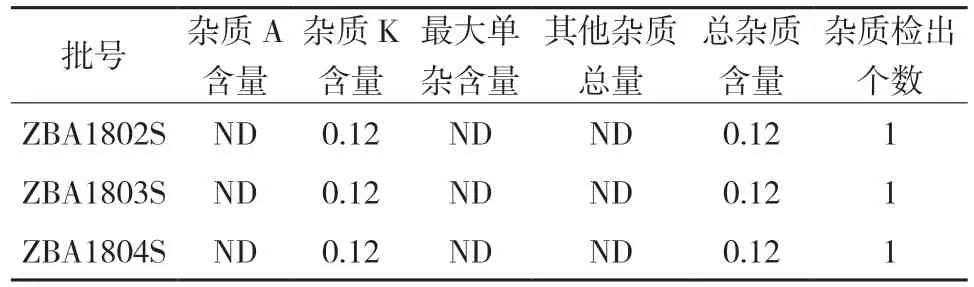
表3 关物质检查结果………… %
3 讨论
据文献报道,河南省药检所于2018年对43批次诺氟沙星胶囊进行了有关物质专项监督抽验,按现行标准检测,部分批次的诺氟沙星胶囊主峰前有一杂质峰与主峰未完全分离,该杂质峰应为杂质E[5];与此同时,浙江省台州市药检所于2017年对57批次诺氟沙星胶囊进行了抽验,有关物质研究结果发现,3个厂家的5批次样品中检出杂质E超标,1批检出杂质K在限度边缘[1]。因此,为有效控制产品质量,诺氟沙星胶囊的有关物质检测方法需要修订提高。
本研究是在《中国药典》收载的诺氟沙星胶囊有关物质检测方法的基础上,对色谱条件进行了调整,经专属性、线性、定量限和检测限、精密度、准确度、耐用性等试验的验证后,确认该检测方法不仅准确可靠,而且可以检测出诺氟沙星的7个杂质,尤其是诺氟沙星原料于合成期间生成的杂质E和杂质K。如表3所示,自研的诺氟沙星胶囊中含0.12%的杂质K,说明该有关物质检测方法更有利于控制原料药及产品的质量。
猜你喜欢
农产品加工(2022年7期)2022-05-26
中国药学药品知识仓库(2022年8期)2022-05-09
河南农业·综合版(2022年2期)2022-03-18
河南农业(2022年2期)2022-03-14
河南农业·综合版(2021年7期)2021-08-23
山东工业技术(2019年6期)2019-03-27
科技创新导报(2017年20期)2017-09-13
领导文萃(2017年6期)2017-03-24
中学生数理化·高一版(2016年7期)2016-12-07
中学生数理化·中考版(2015年12期)2015-09-10
