
Treg调控ILC2参与炎症性疾病的研究进展①
2022-10-02 11:24:18龚展德陈昱丞戴钟玲刘欣袁碧晨罗英雷爱华
中国免疫学杂志 2022年15期
龚展德 陈昱丞 戴钟玲 刘欣 袁碧晨 罗英 雷爱华
(南华大学衡阳医学院病原生物学研究所,衡阳 421001)
二型固有淋巴细胞(group 2 innate lymphoid cell,ILC2)由骨髓中的共同淋巴祖细胞(common lymphoid progenitor cell,CLP)发育分化而来,是近年来发现的一类表面缺乏特异性抗原识别受体的固有淋巴细胞。ILC2表面不表达其他免疫细胞谱系的标志分子,但高表达CD45、CD25、CD127及ST2等表面分子[1]。与Th2类似,ILC2的分化受核心转录因子GATA3调控,且通过分泌二型细胞因子发挥抗寄生虫感染的作用[2-3]。ILC2主要存在于骨髓、肺、肠道及皮肤等器官和组织。在过敏原或感染等因素作用下,上皮细胞受损而产生IL-25、IL-33、胸腺基质淋巴细胞生成素(thymic stromal lymphopoietin,TSLP)等细胞因子。这些细胞因子与ILC2表面相应细胞因子受体结合后,可导致ILC2活化而产生大量IL-5和IL-13,进而参与包括哮喘在内的多种炎症性疾病的发生[1,4-6]。
除细胞因子外,ILC2的产生和活化还受多种物质调控。其中,补体组分C3a、肿瘤坏死因子配体相关分子1A(TNF ligand-related molecule 1A,TL1A)、神经肽(neuromedin U,NMU)、血管活性肠肽(vasoactive intestinal peptide,VIP)、前列腺素D2(prostaglandin D2,PGD2)及嗜酸细胞活化趋化因子等能够促进ILC2的增殖活化;而前列腺素E2(PGE2)、前列腺素I2(PGI2)、IFN-γ、雄激素、儿茶酚胺、丁酸盐等则能够抑制ILC2活化[7]。有趣的是,ILC2通过其表面分子可与其他免疫细胞相互作用,在炎症性疾病的进程中扮演重要角色[8]。例如,ILC2通过其表面表达的MHCⅡ分子可将抗原递呈给T细胞,进而促进T细胞活化[9-10]。ILC2细胞表面的NKP30受体能够通过B7-H6配体直接与皮炎患者皮肤内角化细胞结合活化ILC2细胞[11]。细胞间黏附分子ICAM-1在ILC2的发育与功能中至关重要[12]。值得注意的是,近年来,调节性T细胞(regulatory T cell,Treg)对ILC2的调控作用及其在炎症性疾病中的作用备受关注。
Treg是一类成熟的CD4+T细胞亚群,根据发育的位置不同分为自然Treg(natural Treg,nTreg)和诱导性Treg(inducible Treg,iTreg)。在发育过程中,nTreg需要强烈的CD28共刺激,而iTreg则可在较弱的CD28和CTLA4共刺激下成熟[13]。Treg主要通过细胞接触依赖机制或分泌IL-10和TGF-β发挥免疫抑制作用,因而在炎症性疾病过程中发挥重要作用。例如,在慢性鼻窦炎黏膜中,Treg可显著降低炎症细胞因子和嗜酸性趋化因子水平,并减弱嗜酸性粒细胞(eosinophil,Eos)的募集,进而缓解炎症进展[14]。Treg与Th17间的平衡在炎症性疾病的发生发展中非常重要[15-17]。研究发现在ILC2诱导或参与的炎症性疾病中,Treg可通过调控ILC2反应在疾病的进程中发挥重要作用。下面重点介绍Treg调控ILC2在哮喘、特异性皮炎(atopic dermatitis,AD)及动脉粥样硬化(atherosclerosis,AS)中的作用及相关免疫学机制。
1 Treg调控ILC2在哮喘中的作用
哮喘是一种慢性气道免疫性疾病,涉及多种细胞及细胞因子,以气道高反应性和可逆性气流阻塞、嗜酸性炎症及气道黏膜炎症为特征。目前认为ILC2和Th2通过分泌二型细胞因子(如IL-4、IL-5和IL-13)在哮喘的发生中扮演重要角色。IL-4诱导B细胞产生IgE抗体,IL-5招募并激活Eos,而IL-13可促进黏液产生。ILC2为固有免疫细胞,通常在过敏性疾病的早期发挥作用[18]。有研究发现,活化的ILC2可促进Th2产生[19-21],因而其在哮喘中的作用备受关注。而Treg通过抑制树突状细胞(dendritic cell,DC)和T细胞等细胞功能发挥缓解哮喘的作用[22]。
近年研究发现Treg通过抑制ILC2的活化在缓解过敏性肺炎中发挥重要作用[23-24]。研究发现,在过敏性肺炎小鼠的恢复期,小鼠肺脏中maresin 1含量升高,而外源性给予maresin 1可显著促进小鼠肺脏Treg的扩增;Treg通过分泌TGF-β抑制ILC2的活化,从而缓解肺炎症状[25]。MAAZI等[26]发现人和小鼠的ILC2均表达重要表面分子诱导性共刺激分子(inducible costimulatory molecule,ICOS)和其配体ICOSL;ICOS结合ICOSL介导 了ILC2间 的 相互 作用,并诱导ILC2中STAT5和Bcl-2信号的激活,从而促进ILC2的存活和活化,最终参与过敏性哮喘的发生。有趣的是,iTreg表面表达ICOS分子,亦可与ILC2表面的ICOSL结合[24]。ICOS-ICOSL信号促进了Foxp3+Treg的扩增及其分泌IL-10和TGF-β的能力,而IL-10和TGF-β可显著抑制ILC2分泌IL-5和IL-13的能力,进而缓解ILC2诱导过敏性肺炎的症状[24]。因此,iTreg对ILC2的活化具有较强的抑制作用,在治疗ILC2诱发的哮喘中具有潜在的临床应用价值。
2 Treg调控ILC2在AD中的作用
近年研究发现ILC2与AD的发生密切相关。在AD患者体内,ILC2细胞数量明显增多,并可通过产生IL-5导致Eos聚集,进而引发炎症[27-28]。同时,皮肤中的肥大细胞及嗜碱性粒细胞可分泌IL-4及PGD2作用于ILC2表面相应受体进而促进ILC2活化,促进炎症的发生[28-29]。更为重要的是,皮炎中的ILC2亦受Treg的调控。研究发现,皮肤驻留RORα+Treg可抑制ILC2活化,进而缓解过敏性皮炎[30]。RORα+Treg和ILC2表面均表达死亡受体3(death receptor 3,DR3)分子;其可与内皮细胞或髓系细胞来源的TL1A结合,进而增强Treg功能,与ILC2表 面DR3结 合 亦 可 激 活ILC2。因 此,RORα+Treg和ILC2对TL1A存在竞争关系。靶向敲除皮肤驻留Treg中RORα的小鼠在经MC903诱导发生AD后,ILC2来源的IL-5含量明显增加,Eos数量上升,二型免疫反应增强,AD症状加重。这是由RORα缺失的Treg表面DR3表达水平下降,无法与ILC2竞争TL1A所致[30]。此外,与人外周血中的Treg相比,人皮肤Treg表达更高水平的RORα[30],提示RORα+Treg在抑制由TL1A/ILC2诱导的AD中可能发挥重要作用。
3 Treg调控ILC2在AS中的作用
AS是一种由脂质驱动的慢性血管炎症,其发生始于低密度脂蛋白在血管壁的过度积累;随后脂质在血管上氧化堆积,致使内皮受损,进而导致血管免疫调节失衡。AS的发生涉及多种炎症细胞及炎症介质的相互作用。作为具有负向免疫调节作用的细胞,Treg在AS中的作用主要体现在以下两方面:一方面,Treg能下调血清中的胆固醇等相关脂质含量水平;另一方面,Treg能以细胞接触的方式或通过分泌抑制性细胞因子IL-10和TGF-β以减少相关炎症细胞的浸润与炎症因子表达[31-33]。同时,研究发现ILC2在AS的发病过程中也发挥保护作用。活化的ILC2可通过分泌IL-5促进B1细胞产生天然抗磷脂酰胆碱的IgM;其在AS的早期预防中发挥重要作用。此外,过继转移ILC2可提高M2巨噬细胞水平,降低AS斑块中的脂质含量,减少斑块形成。
最新一项研究表明,Treg可通过促进ILC2的活化共同对抗AS[34]。该研究的体内外实验发现,CD4+Foxp3+Treg可通过分泌TGF-β和IL-10与细胞接触的方式促进ILC2增殖及其分泌IL-13的能力。过继转移Treg增加主动脉旁淋巴结和脾脏中ILC2比例和数量,减少了主动脉斑块产生及斑块中巨噬细胞浸润,延缓了AS进展[34]。ILC2经Treg活化产生高水平的IL-13是否通过促巨噬细胞向M2极化进而达到抗炎作用还需进一步探究。但与哮喘中Treg对ILC2的抑制作用不同,AS中Treg促进了ILC2的活化。此项研究提示,不同疾病不同组织中ILC2的生物学特性及功能不尽相同,而AS与哮喘小鼠肺脏中Treg-ILC2轴的调控分子机制差异尚待阐明。
4 结语
综上所述,ILC2在炎症性疾病的发生发展中发挥重要作用。Treg作为具有免疫抑制功能的CD4+T细胞,通过与ILC2的直接接触或释放TGF-β和IL-10在抑制ILC2功能及缓解ILC2诱导哮喘和特异性皮炎的过程中发挥重要作用;而在AS中,Treg可促进ILC2的活化,减缓AS病程(图1)。此外,研究发现在一些自身免疫病中,ILC2亦可通过调控Treg缓解疾病进程。例如,在慢性关节炎中,ILC2通过分泌IL-9导致Treg活化,从而促进炎症的消退[35]。因此,ILC2与Treg间的相互作用较为复杂。在不同炎症性疾病中,Treg与ILC2间的调控作用不尽相同。进一步深入研究Treg与ILC2间的调控机制不仅有利于炎症性疾病发病机制的阐明,还可为炎症性疾病的治疗提供新策略,具有重要的理论和临床意义。
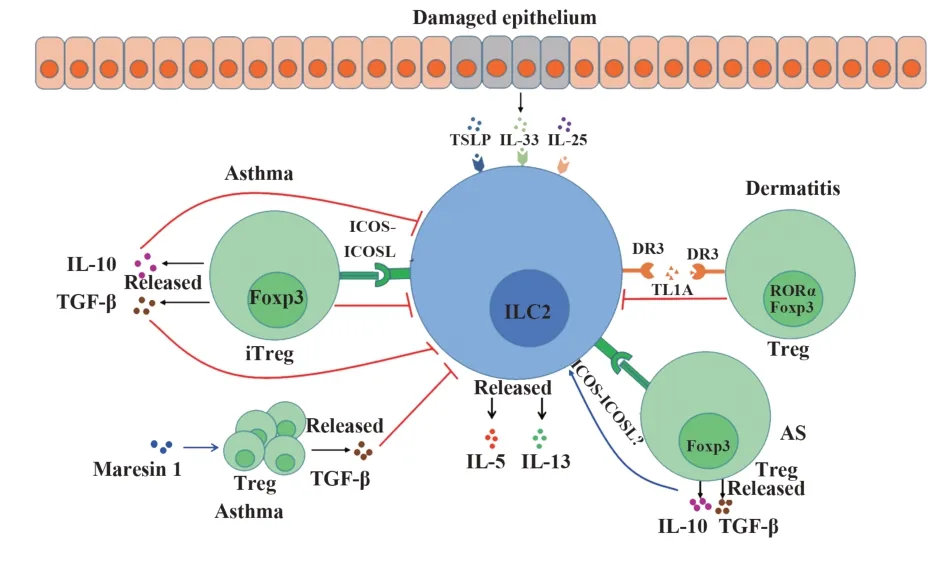
图1 Treg在不同炎症性疾病中对ILC2的调控作用Fig.1 Modulation of ILC2 by Treg in different inflammatory diseases
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10 09:15:38
中老年保健(2021年8期)2021-08-24 06:22:06
现代临床医学(2021年4期)2021-07-31 07:55:54
作文成功之路·小学版(2020年6期)2020-07-27 01:48:28
医学新知(2019年4期)2020-01-02 11:03:52
医学新知(2019年4期)2020-01-02 11:03:52
中西医结合心脑血管病杂志(2016年20期)2016-03-01 04:20:37
医学研究杂志(2015年12期)2015-06-10 06:57:46
无机化学学报(2014年10期)2014-02-28 17:33:13
河南医学研究(2014年4期)2014-02-27 14:52:16
