
蛋白冠的形成、分析及生物效应研究进展
2022-09-28 07:48汤杰张相朱娜丽李灵香玉王亚韡
生态毒理学报 2022年3期
汤杰,张相,朱娜丽,李灵香玉,*,王亚韡
1.国科大杭州高等研究院环境学院,杭州 310024
2.中国科学院生态环境研究中心环境化学与生态毒理学国家重点实验室,北京 100085
纳米颗粒被生物摄入到体内后容易与蛋白质相互作用。此时,蛋白质自发的吸附到纳米颗粒表面,形成纳米颗粒-蛋白质的核壳结构复合物,其表面蛋白质被称作“蛋白冠”。根据蛋白质与纳米颗粒表面亲和力的差异,蛋白冠被分为“硬冠”和“软冠”[1]。当蛋白质开始与纳米颗粒相互作用时,软冠借助弱相互作用吸附在纳米颗粒表面,此时溶液中蛋白质和吸附在纳米颗粒表面的蛋白质处于动态平衡状态。随后发生Vroman效应[2],即最初的高浓度及具有高迁移率但是亲和力相对低的蛋白质将逐渐以弱结合的低稳定态吸附在纳米颗粒表面,随着时间的推移,这些低亲和力的软冠蛋白会被亲和力更强的蛋白质取代[3],进而改变蛋白冠组成,形成硬冠。通常,软冠被描述为一种高度动态且松散的蛋白质层,而硬冠比软冠具有更高的结合能,使得硬冠在纳米颗粒表面的吸附不可逆[4-7]。
蛋白冠的形成使得纳米颗粒具有新的生物学特征,并影响纳米颗粒在生物体或环境系统中的吸收/吸附、分布、转化和归趋等[8]。这些特征对纳米颗粒的生物效应[9]和靶向纳米药物的设计[10-12]等都具有重要意义。因此,在过去的10多年中,蛋白冠已成为一个研究热点,由于蛋白质易于分析和结构表征,而且蛋白质在受体参与和信号传递中起着关键作用,因此近些年的研究的重点和核心主要在蛋白冠上。
本文基于近期的文献资料,较为系统地对蛋白冠的形成及影响因素、蛋白冠的表征分析方法和蛋白冠对纳米颗粒生物效应的影响进行归纳总结,并展望了后续的重点研究方向,以期待对蛋白冠的形成、应用和风险评估研究提供一定的参考。
1 蛋白冠的形成(Formation of protein corona)
1.1 蛋白质吸附的作用机制
纳米颗粒被生物体摄入后,会被体内游离的蛋白质所包围。蛋白质通过扩散或沿着势能梯度迁移到纳米颗粒表面,不断地相互竞争纳米颗粒表面的结合位点,以吸附到纳米颗粒上形成蛋白冠。文献资料表明,蛋白冠的形成主要由库仑力和范德华力、氢键和疏水相互作用等介导[8,13-14]。热力学因素影响蛋白冠的形成,只有在热力学有利的条件下,蛋白质吸附才会自发地发生。首先,高丰度蛋白质会短暂形成软蛋白冠,随着时间的增加,根据Vroman效应,高亲和力蛋白质会逐渐取代软蛋白冠吸附到纳米颗粒表面,进而形成硬蛋白冠,并最终在纳米颗粒表面形成单层或多层的蛋白冠结构。蛋白质对纳米颗粒的亲和力决定了它们与其他物质发生相互作用或转移到新的流体介质时的行为与归趋是吸附、保持结合还是解离[14]。蛋白冠的结合是一个自发的过程(公式(1)[14]),在整个过程中焓降低,同时纳米颗粒周围的水合层被取代,也导致了熵的增加。其中ΔGads、ΔHads和ΔSads分别表示吸附过程中吉布斯自由能、焓和熵的变化,T表示温度。
ΔGads=ΔHads-TΔSads<0
(1)
共价键和非共价键的形成、界面水分子的重排以及蛋白质或纳米颗粒表面的构象变化等一系列反应都会使焓减少(ΔHads<0)或熵增加(ΔSads>0),这些均有利于蛋白质在纳米颗粒表面的吸附。吸附过程中所涉及的作用机制取决于蛋白质和纳米颗粒的物理化学性质。
吸附过程中ΔGads决定了蛋白冠-纳米颗粒复合物的稳定性。具有较大ΔGads的蛋白冠解吸释放到孵育溶液的概率较低,且倾向于和纳米颗粒保持结合。而有着较小ΔGads的蛋白冠很容易解吸并释放到孵育溶液中。由此,带电荷或疏水的纳米颗粒比亲水性的纳米颗粒更易与蛋白质形成稳定的相互作用[14]。
总体上,蛋白质与纳米颗粒相互作用形成蛋白冠的作用机制包括非极性氨基酸的疏水作用、共价键结合、范德华力、蛋白质与蛋白质的相互作用、静电吸引或排斥作用、阳离子桥连作用、配体交换、氢键作用、蛋白质与其他生物分子的螯合或置换作用等[8](图1)。资料显示,部分相互作用可能导致吸附在纳米颗粒表面的蛋白质变性,进而影响蛋白质结构和功能[15]。
在蛋白冠形成过程中,蛋白质可能发生结构重排,即“构象变化”。如果蛋白质内的疏水或带电序列分别与疏水或带电的纳米颗粒表面相互作用,则有利于构象的热力学变化,大多数蛋白冠在对纳米颗粒的吸附过程中都表现出了一定程度的构象变化[16]。然而,构象变化的程度取决于蛋白质和纳米颗粒的化学与结构特征。具体表现为:具有较强内部稳定性(如盐桥或二硫键[17])的蛋白质在吸附过程中通常会发生较小的构象变化,相反,带电或疏水的纳米颗粒比亲水性的纳米颗粒更能改变吸附蛋白质的构象。有研究探讨了牛血清白蛋白(bovine serum albumin,BSA)与石墨烯碳基修饰的单壁碳纳米管(single-walled carbon nanotubes,SWNTs)之间的相互作用[18]。结果显示,BSA蛋白冠作为一个弱受体,可以促进电荷在SWNTs结构中转移;但是由于SWNTs尖锐和分立的电子态密度,电荷转移只发生在SWNTs,却不发生在石墨烯中。而且,BSA二级结构中外α螺旋的弛豫随电荷的转移而增加[19]。蛋白质构象的改变通常是不可逆的,包括在解吸之后。例如,人转铁蛋白暴露于超顺磁氧化铁纳米颗粒后,铁的释放改变了该蛋白的主要功能,但磁性纳米颗粒去除后,原蛋白的构象并没有恢复,转铁蛋白的构象已经从紧密结构变为开放结构,发生了不可逆的变化[18]。
蛋白质-蛋白质之间相互作用可以调节这些结构的变化,而且它们也可以导致已吸附的蛋白质被亲和力更强的蛋白质所取代。此外,蛋白质吸附会增强纳米颗粒之间的相互作用,使其产生团聚,团聚物的形成会对纳米药物的有效性产生不利影响[13]。在复杂生理环境中,蛋白冠的形成涉及到许多蛋白质通过蛋白质-纳米颗粒和蛋白质-蛋白质之间相互作用。每一种相互作用都有其特有的结合能。有一种流行的假设是,硬蛋白冠直接与纳米材料表面相互作用,而软蛋白冠通过弱蛋白质-蛋白质与硬冠蛋白之间相互作用。另一种假设是硬冠和软冠中的蛋白质都可以与具有不同结合能的纳米颗粒表面直接相互作用。然而,目前几乎没有实验证据来区分以上2种假设[14]。
1.2 蛋白质的吸附动力学
研究表明,蛋白冠的特性及其相对丰度在本质上是动态变化的[20]。对于特定类型的纳米颗粒,蛋白冠的动态特征主要受以下因素影响:(1)不同蛋白质吸附在纳米颗粒表面的亲和力和平衡常数的差异非常大,导致不同蛋白质在纳米颗粒表面的停留时间不同;(2)纳米颗粒-蛋白质结合/解离常数的变化;(3)生物体内蛋白质种类的差异。总的来说,蛋白冠的形成是蛋白质在纳米颗粒表面上连续吸附和解吸平衡的动态过程,其中的结合常数(kon)和离解常数(koff)所描述的动力学过程很重要(图1)。kon的值取决于蛋白质与纳米颗粒的接触频率,koff的值取决于蛋白质与纳米颗粒复合物的结合能。kon和koff这2个常数之间的平衡决定了蛋白质与纳米颗粒的亲和力,并将其定义为解离常数(Kd)。目前,通过表面等离子体共振(SPR)或石英晶体微天平(QCM)等技术,根据Langmuir吸附等温线或Scatchard方程,可以得到实时的定量吸附曲线,并掌握蛋白冠形成的Kd。例如,血清蛋白在纳米颗粒上吸附的Kd大约在10-4~10-9mol·L-1之间,与抗体-抗原之间相互作用的Kd范围相似[21]。
吸附动力学与时间有关,其速率与蛋白质浓度和Kd有关。然而,当介质是生理溶液时,在复杂混合物中测量每个蛋白质的kon和koff来估算Kd值的方法仍有难度。作为替代方法,可以用双指数函数来建模[22]。这个模型将蛋白质吸附和解吸分为“快”和“慢”2个部分,每一部分都有自己的“有效”kon和koff(图1)。吸附和解吸的快和慢组分分别代表硬冠和软冠。文献资料显示,在血浆蛋白吸附到N-异丙基丙烯酰胺/N-叔丁基丙烯酰胺共聚物纳米颗粒过程中,快组分(硬冠)在几秒钟内形成,而慢组分(软冠)则在几分钟到几小时的时间范围内才形成,其解吸行为都比较相似,快组分(软冠)的平均寿命约为10 min,慢组分(硬冠)的平均寿命约为8 h[23]。类似的动力学行为也适用于血浆蛋白在其他纳米颗粒表面的吸附。综上,软蛋白冠和硬蛋白冠的概念模型及介导蛋白冠形成的物理化学相互作用如图1所示。
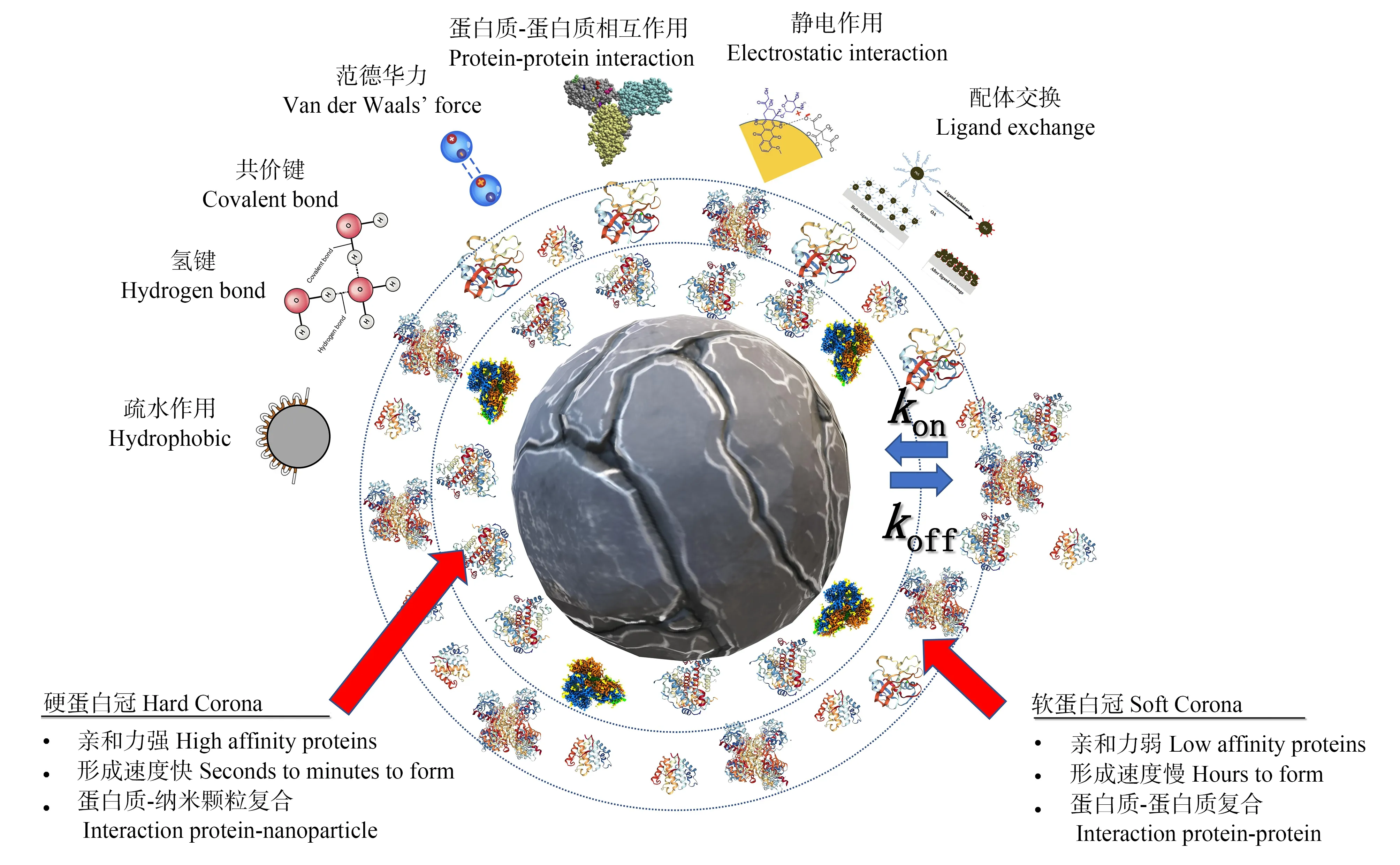
图1 软蛋白冠和硬蛋白冠的概念模型及介导蛋白冠形成的物理化学相互作用Fig. 1 Conceptual model of soft and hard protein corona and physicochemical interactions that mediate protein corona formation
1.3 蛋白冠形成的影响因素
1.3.1 纳米颗粒的物理化学属性
各种各样的纳米颗粒由许多不同的材料合成,具有不同的界面性质,这些性质影响纳米颗粒与生物分子的相互作用[13]。针对蛋白质与不同材料纳米颗粒之间相互作用的研究已经开展较多,证实了表面自由能、颗粒形貌、尺寸、电荷和疏水性等因素对蛋白质吸附过程的影响[24]。
蛋白质在纳米颗粒表面吸附的结合常数和结合位点数很大程度取决于纳米颗粒的尺寸和形貌。研究发现,蛋白质在与纳米颗粒结合过程中经历了结构变化,其构象变化在颗粒尺寸较大的情况下显得更为明显[25]。此外,纳米颗粒的形态转化,如氧化、硫化、还原和溶解等非生物或生物转化过程,能够剧烈影响纳米颗粒的表面形貌,甚至实现对颗粒表面的修饰这些变化均能影响纳米颗粒表面的蛋白冠形成与动态变化。
蛋白质与纳米颗粒之间的相互作用受纳米颗粒的表面特性影响。无机纳米颗粒多为小晶体,其表面性质取决于暴露的晶面、边缘和漩涡等,表面缺陷的存在会导致纳米颗粒结构不稳定从而容易发生重构。因此,结晶型纳米颗粒表面通常有配体修饰,如羧基、胺或其他基团,使其具有化学和胶体稳定性。研究表明纳米颗粒的表面修饰能够影响蛋白冠的形成:聚丙烯酸酯聚合物修饰的TiO2纳米颗粒主要借助表面羧基和氨基酸羟基的酯化作用,通过该共价键形成蛋白冠。与此同时,TiO2表面修饰的羧基和蛋白质正电荷也可通过静电作用形成蛋白冠。而对于聚乙烯吡咯烷酮表面修饰的TiO2,其表面为电中性,因此其主要借助氨基酸与聚乙烯吡咯烷酮之间的氢键和范德华力吸附氨基酸形成蛋白冠[26]。
聚合物纳米颗粒是一类具有高度通用性的纳米材料,具有出色的生物相容性,在临床医疗方面表现出优良性能[27-28]。它们由疏水的core-forming聚合物和shell-forming聚合物(一般为带电或亲水的)组成,可被靶向或蛋白排斥基团功能化。针对血清蛋白与功能化聚合物纳米颗粒的相互作用研究发现,带正电的氨基-纳米颗粒通过静电相互作用,对血清蛋白表现出增强的吸附作用,形成由多种蛋白质成分组成的硬蛋白冠。但是亲脂性物质不断从疏水纳米颗粒中滤出,所以蛋白质在颗粒表面的吸附明显改变了纳米颗粒的稳定性,严重损害了纳米颗粒的完整性。这些结果强调了表面修饰对聚合物纳米颗粒形成蛋白冠的影响不容忽视,尤其在纳米医药应用领域,蛋白分子的吸附可能会改变胶体药物传递系统的完整性和时效性,进而削弱其性能[29]。所以,在纳米尺度上对纳米颗粒的物理化学属性进行精确的表征对进一步推进蛋白冠研究至关重要。
1.3.2 蛋白质的性质
蛋白质是由20种不同氨基酸组成的线性聚合物链,在适当条件下(通常是自发地)折叠成特定的具有生物活性的3D结构,其存在很大的内在结构复杂性,使得蛋白质具有不同的生物功能。蛋白质的热力学稳定性不高,数千个原子组成的小蛋白质的稳定性只相当于几个氢键[30]。而且,蛋白质之间的热力学稳定性可能存在很大差异,从而影响它们在纳米颗粒表面上的结构响应[31]。
纳米颗粒暴露在生物环境中,倾向于和蛋白质分子相互作用,形成蛋白冠。蛋白冠赋予纳米颗粒独特的生物学特性,其蛋白质组成对纳米颗粒的生物学命运起决定性作用。蛋白质的生理行为源于其物理化学性质,包括表面电荷、疏水性和结构稳定性等。有研究假设蛋白质的物理化学性质会影响它们与纳米颗粒的相互作用,并对纳米颗粒与细胞的相互作用具有决定性的影响[32]。Dhar等[33]使用不同结构类型的蛋白质模型来研究蛋白质二级结构对纳米颗粒-蛋白质界面的影响。结果显示,蛋白质的表面电荷决定了纳米颗粒-蛋白质之间一次相互作用的性质和二次相互作用的程度,从而导致蛋白质结构重排,同时,蛋白质二级结构的含量对纳米颗粒-蛋白质界面的二级相互作用程度和纳米蛋白复合物的分散状态都存在显著影响。显然,蛋白质的物理化学特性在蛋白冠形成过程中对纳米颗粒-生物界面的调节起着重要作用。
1.3.3 介质条件
蛋白质和纳米颗粒的相互作用受介质环境的各种因素影响,如温度、pH、盐度和流动性等。环境pH值的变化会改变蛋白质的性质从而影响蛋白质与纳米颗粒的结合。针对生物偶联物在宽泛pH范围的缓冲水溶液中的行为研究发现,生物偶联物在中性和碱性溶液中是稳定的,其在pH低于蛋白质等电点时发生聚集,且蛋白质的二级结构在颗粒稳定的条件下保持不变;但当生物偶联物聚集时,蛋白质的构象发生了一些微小变化[34]。该结果表明,保持蛋白质结构与纳米颗粒相互作用的重要性,以驱动生物偶联物的稳定性。有研究表明,轻微的温度变化,如人类体温随昼夜更替的改变,会影响蛋白质的亲和力和蛋白质-纳米颗粒复合物的细胞摄取或毒性[35]。动态条件下血浆蛋白质吸附及表面诱发血栓的研究表明,在静态和流动条件下,聚N,N-二甲基丙烯酰胺(poly N,N-dimethylacrylamide,PDMA)修饰表面对血浆蛋白的吸附以及对血小板粘附程度均低于其他表面修饰,而且吸附蛋白冠的分布与测试条件(静态或流动)以及聚合物表面修饰的化学特性有很强的相关性[36]。近期,有研究者考察了pH和盐度对肌红蛋白在二氧化硅纳米颗粒上形成蛋白冠的影响[37]。结合吸附结果和肌红蛋白的表面静电映射,证实了在低盐浓度下能形成单层硬蛋白冠,当盐不断加入后单层硬冠逐渐转变为多层的硬冠和软冠。进一步机理研究显示,蛋白质吸附行为随pH和盐度增加的变化主要归因于静电相互作用和表面电荷调节效应的改变。所以,介质条件对蛋白冠形成的影响研究有助于揭示蛋白质在纳米颗粒表面的吸附机理。
2 蛋白冠的组成和结构分析(Characterization of protein corona composition and structure)
蛋白冠的组成和结构分析主要包括厚度和密度、特性和数量、排列和取向、构象及亲和力等重要参数。其中,蛋白冠的厚度和密度决定了纳米-蛋白复合物的总体尺寸和优先暴露的表面;吸附在纳米颗粒表面的蛋白质的特性和数量决定了一系列可能发生的生化反应及其反应强度;蛋白冠排列和取向则影响了潜在的结合域和催化域;而蛋白冠构象影响蛋白质的活性及其与其他生物分子的相互作用[14]。
显然,蛋白冠组成和结构的分析表征对于深入研究蛋白冠形成机理及预测其后续行为特征十分重要。现有分析技术大致可分为两大类:原位和非原位检测技术。原位测量是一种具有挑战性的分析蛋白冠的方法,因为分析过程中无需与周围介质分离,所以不影响蛋白冠的成分和动态平衡。当蛋白冠处于特定生理条件下时,原位检测技术能更直接地分析表征出蛋白冠的实时动态信息。然而,由于分析技术的瓶颈,通过原位测量获得的信息量仍然是有限的。所以大多数研究依然通过非原位检测技术,将蛋白冠-纳米颗粒复合物与未结合的游离蛋白质分离,进而开展分析表征。
2.1 蛋白冠的原位检测技术
原位检测可以直接在介质中分析吸附在纳米颗粒表面的蛋白冠,从而不影响蛋白冠的成分,反映出蛋白冠最自然的状态。但是介质中除了与纳米颗粒结合的蛋白冠外,还存在游离的蛋白质,可能干扰分析的准确性,成为蛋白冠原位分析技术发展的瓶颈。例如,动态光散射(dynamic light scattering,DLS)可以确定样品的表观扩散系数,通过斯托克斯-爱因斯坦方程能计算出颗粒物水合半径[38],也可以通过测量时间-粒径分布的关系反映蛋白冠稳定性。与DLS类似,荧光相关光谱法(fluorescence correlation spectroscopy,FCS)可以在亚纳米精度下,通过蛋白质浓度的函数计算颗粒物水合半径,借此实现蛋白冠-纳米颗粒复合物尺寸的原位分析,用于表征蛋白冠尺寸[39-40]。与DLS相比,FCS只检测荧光标记物,介质中其他成分不会干扰荧光信号,因此不需要分离游离的蛋白质。但FCS的缺点也非常明显,其需要使用荧光标记的纳米颗粒和蛋白质,而引入标记可能会引发系统本身发生变化,从而影响蛋白冠表征结果[41]。此外,蛋白质吸附在纳米颗粒表面通常会导致蛋白质荧光猝灭,因此借助荧光猝灭法(fluorescence quenching,FQ)可以分析蛋白质与纳米颗粒之间的相互作用[42]。
目前,流式细胞技术结合荧光标记抗体是一种原位检测蛋白冠的中特定蛋白成分的新兴技术[43]。通常,纳米颗粒吸附蛋白质后由于质量的增加,其布朗运动也会减慢,因此可以通过纳米颗粒跟踪分析法(nanoparticle tracking analysis,NTA)测量蛋白冠尺寸的动态变化[44]。最近通过对纳米颗粒标记同位素,借助核磁共振光谱(nuclear magnetic resonance,NMR)测量颗粒物扩散系数,经由颗粒物水合半径的增加来量化纳米颗粒表面吸附形成的蛋白冠,从而实现原位表征蛋白冠,因该方法不基于光学技术,所以可以在富含细胞等浑浊的生物体液中原位分析蛋白冠[45-47]。
2.2 蛋白冠的异位检测技术
2.2.1 蛋白冠分离
一般来说,分离会改变吸附蛋白质和游离蛋白质之间的平衡,因此所有分离技术都会对蛋白冠的组成和结构产生一定的影响。在大多数分离过程中,软蛋白冠将被去除,所以对其组成、结合方式和生物相关性知之甚少。目前,经过分离、洗涤步骤后,仍吸附在纳米颗粒上的蛋白质在分析表征时被定义为硬蛋白冠。由于存在许多分离游离蛋白质的方法,因此,在对硬冠和软冠的研究中精确定义硬-软冠边界以及2层之间的区别存在巨大的挑战[48]。有研究表明,有些蛋白质无法用常规方法从纳米颗粒表面分离,因此部分硬蛋白冠很难被全面分析。甚至有研究把这些特别紧密结合的蛋白质称为“界面蛋白冠”[49]。
离心是将带有蛋白冠的纳米颗粒与周围介质分离的最常用方法,其原理是将纳米颗粒-蛋白质复合物颗粒化,从而分离未结合的蛋白质。仅仅通过第一次离心,结合松散的蛋白质很可能仍附着在纳米颗粒表面,所以样品在相应的缓冲液中需要经历多次洗涤。通过多次洗涤步骤,蛋白冠的平衡会重新调整,结合松散的蛋白质在进行分析之前会被完全去除。迄今为止,离心可能是分离强结合蛋白质最成熟的方法[5]。如果样品在离心过程中稳定,只需要通过样品与周围介质的密度差异就可以分离,但在离心的过程中蛋白冠与介质的接触面积会因为纳米颗粒-蛋白质复合物沉积团聚而减少,为了避免这种情况,也可以在添加蔗糖垫的条件下进行离心[50-51],这样可避免样品和介质之间的不确定接触。离心分离的操作必须非常小心,因为高丰度的蛋白质聚集体很容易与纳米载体-蛋白质复合物一起沉淀,可能会被误认为是蛋白冠的一部分。磁性分离是另一种分离蛋白冠-纳米颗粒复合物的有效方法,但其仅适用于有磁性的纳米颗粒[52-55]。基本的分离过程是通过磁铁靠近样品,把纳米颗粒集中在磁铁附近,从而去除上清液。通过在缓冲液中重新悬浮,重复该操作过程来洗涤样品。离心和磁分离这2种方法有一定的相似之处,它们都依赖于重复的洗涤步骤来去除多余的蛋白质,每次清洗样品都会达到新的平衡,因此蛋白冠很大程度被相应的洗涤程序所影响。
除了上述2种基于密度和磁性差异的分离方法,借助流体动力学尺寸差异,也可以通过色谱法和类似色谱法的技术进行分离。其中,非对称流场流分离技术(asymmetrical flow field-flow fractionation,AF4)作为一种类似色谱的技术,通过流体与外场的共同作用,利用样品扩散系数的差异以实现分离。AF4没有固定相,是一种非常温和的分离技术,适用于分离蛋白质聚集体和蛋白质-纳米颗粒复合物等样品[11,56-58]。目前,也存在另一种类似色谱的技术即流体动力色谱(hydrodynamic chromatography,HDC)用于蛋白冠分离,其工作原理与AF4相似,根据流体动力学尺寸分离样品。在毛细管或填充柱内,通过压力驱动以抛物面流型前进,由于大分子的排斥效应大于小分子,因此它们将更快地移动并优先洗脱[59-60]。尺寸排阻色谱(size exclusion chromatography,SEC)也是分析领域常用的分离技术,这项技术根据样品的流体动力学尺寸分离样品[61-63]。这种方法的缺点是样品和色谱柱填料之间存在剪切应力,这不仅会增加流速,而且可能去除某些结合松散的蛋白质,从而改变蛋白冠的组成[3]。上述3种方法均基于流体动力学尺寸差异进行分离。还有一种分离技术即毛细管电泳(capillary electrophoresis,CE),其原理则是基于电荷比的大小,通过电场使样品电泳从而进行分离,是蛋白冠分离领域的一项有前景的技术,分离后的复合物用于进一步分析[64-67]。与AF4和HDC相同,CE不存在固定相,是一种相对温和的分离技术。
2.2.2 直接检测技术
研究表明,借助透射电子显微镜(transmission electron microscopy,TEM)和扫描电子显微镜(scanning electron microscopy,SEM)可以直接观察到吸附在纳米颗粒表面的蛋白冠[68-69],从而验证蛋白冠存在与否,但该方法不能实现定量分析。如果需要对吸附蛋白量进行定量分析,则可利用标准生化蛋白实现定量测定,比如考马斯亮蓝(Bradford)法[70-71]或二喹啉甲酸(bicinchoninic acid,BCA)法[72-74]。为进一步研究蛋白冠组成,蛋白质通常需要从纯化后的蛋白质-纳米颗粒复合物中分离解吸。目前,分离手段主要根据蛋白冠和纳米颗粒表面互相作用的强度进行选择,通常采用高温、高盐、去污剂或蛋白酶等方法。十二烷基硫酸钠聚丙烯酰胺凝胶电泳法(SDS-polyacrylamide gel electrophoresis,SDS-PAGE)通过将蛋白质凝胶图与已知标准物质比对,以此获得蛋白冠组成[75-78]。SDS-PAGE具有使用广泛、价格低廉和操作简单等优点,但也存在通量低、灵敏度低和伪影等缺点。后续研究发现液相色谱串联质谱(liquid chromatography tandem mass spectrometry,LC-MS/MS)比SDS-PAGE具有更高的通量、准确度和灵敏度,可以实现对蛋白冠组成成分的定量分析,其与SDS-PAGE串联使用可同时实现蛋白冠的定性和定量分析[79-81]。近期研究发现,使用SDS-PAGE切割条带消解后联用LC-MS/MS的分析方法也存在一定缺陷,主要表现在颜色弱或基本没颜色的蛋白质凝胶条带容易被忽视,而这其中可能包含着一些具有重要生理功能的蛋白[82]。此外,该方法也无法提供蛋白冠的蛋白结构信息。傅里叶红外光谱法(Fourier transformed infrared spectrometry,FT-IR)和圆二色谱(circular dichroism,CD)是比较常见的测量蛋白冠构象的分析技术。FT-IR对与酰胺键对应的振动带位移和形状变化非常敏感,可以观察到蛋白质的二级结构变化,能够测量硫蛋白冠与纳米颗粒结合随时间的变化趋势[17]。CD能够确定蛋白质的二级结构变化[83]。表面增强拉曼光谱(surface enhanced Raman scattering,SERS)可以提供金属纳米颗粒表面附近分子的结构、组成和相互作用信息,从而实现对蛋白冠的表征[84]。此外,原子力显微镜(atomic force microscopy,AFM)[85]和FQ[86]也可以检测蛋白冠的结构变化。
2.2.3 间接检测技术
间接检测技术通过测量蛋白冠-纳米颗粒的特性变化来分析蛋白冠,例如尺寸大小、电荷、密度、质量、吸光度和荧光等特性。通常,异位检测技术有凝胶电泳(gel electrophoresis,GE)和差示离心沉降法(differential centrifugal sedimentation,DCS),它们通过矩阵中蛋白冠-纳米颗粒复合物的移动来推断其尺寸大小。GE主要借助外加电场实现带电颗粒物在凝胶孔中迁移。当蛋白冠存在时,由于尺寸增加,蛋白冠-纳米颗粒在凝胶上迁移速度减缓,导致颗粒物迁移速率根据尺寸/电荷比的不同而不同[87]。此外,通过重力或压力驱动的凝胶孔(如SEC),可以对不同尺寸的蛋白冠-纳米颗粒复合物进行分离。对于DCS,沉降时间是纳米颗粒大小和密度的函数,基于纳米颗粒的平均密度,可以确定颗粒物的水合粒径[88-89]。所以DCS已在蛋白冠尺寸检测中被广泛应用,但该技术的蛋白冠厚度的测量值通常偏低。
激光多普勒测速法(laser Doppler anemometry,LDA)和可调谐电阻脉冲传感技术(tunable resistive pulse sensing,TRPS)可检测颗粒物表面电荷的变化。LDA通过激光照射纳米颗粒,使其向相反电荷的电极迁移,根据迁移速率检测ζ电位,而蛋白冠会改变ζ电位,且其受纳米颗粒初始电荷、蛋白质特性和介质pH值等因素影响[90]。ζ电位也可通过TRPS测量。TRPS是一种用于表征缓冲液、血清和血浆中ζ电位的简单且快速的检测方法,可检测颗粒表面蛋白质的变化、大小和分布等[91-92]。金属纳米颗粒在紫外-可见-近红外光谱范围内具有局域表面等离子体共振(localized surface plasmon resonance,LSPR)特征,纳米颗粒表面蛋白冠的形成通常会导致等离子体峰的红移和变宽,因此紫外-可见(UV-Vis)吸收光谱被应用于表征等离子体纳米颗粒上蛋白冠的吸附。研究显示,LSPR频率对纳米颗粒周围环境的微小变化非常敏感[93-94]。利用UV-Vis光谱检测可以杂化纳米颗粒等离子体消光的变化,同时表征蛋白冠的关键参数厚度和折射率[95]。如所测纳米颗粒不是等离子体,也可与表面等离子体共振(surface plasmon resonant,SPR)装置结合进行检测,该技术通过SPR峰角位置的变化来检测蛋白质与纳米颗粒的结合[96]。
石英晶体微天平(quartz crystal microbalance,QCM)和悬浮微通道谐振器(suspended microchannel resonator,SMR)能够测量蛋白冠形成所引起的质量变化,都具有很高的灵敏度。其中,QCM需要将纳米颗粒固定在石英表面,记录蛋白冠形成的质量,实现对蛋白冠的定量分析[97],而SMR则测量谐振腔内微通道的纳米颗粒悬液质量[98]。此外,等温滴定量热法(isothermal titration calorimetry,ITC)能测量纳米溶液在注射蛋白质后热量随时间的变化,积分后构建热量与蛋白冠-纳米颗粒复合物的摩尔比相关函数,以确定蛋白冠形成的热力学参数,如亲和常数和结合化学计量数等信息,以此作为一种补充方法,加深我们对蛋白冠-纳米颗粒复合机制的理解[99]。综上,蛋白冠结构和组成的表征技术的总结如表1所示。
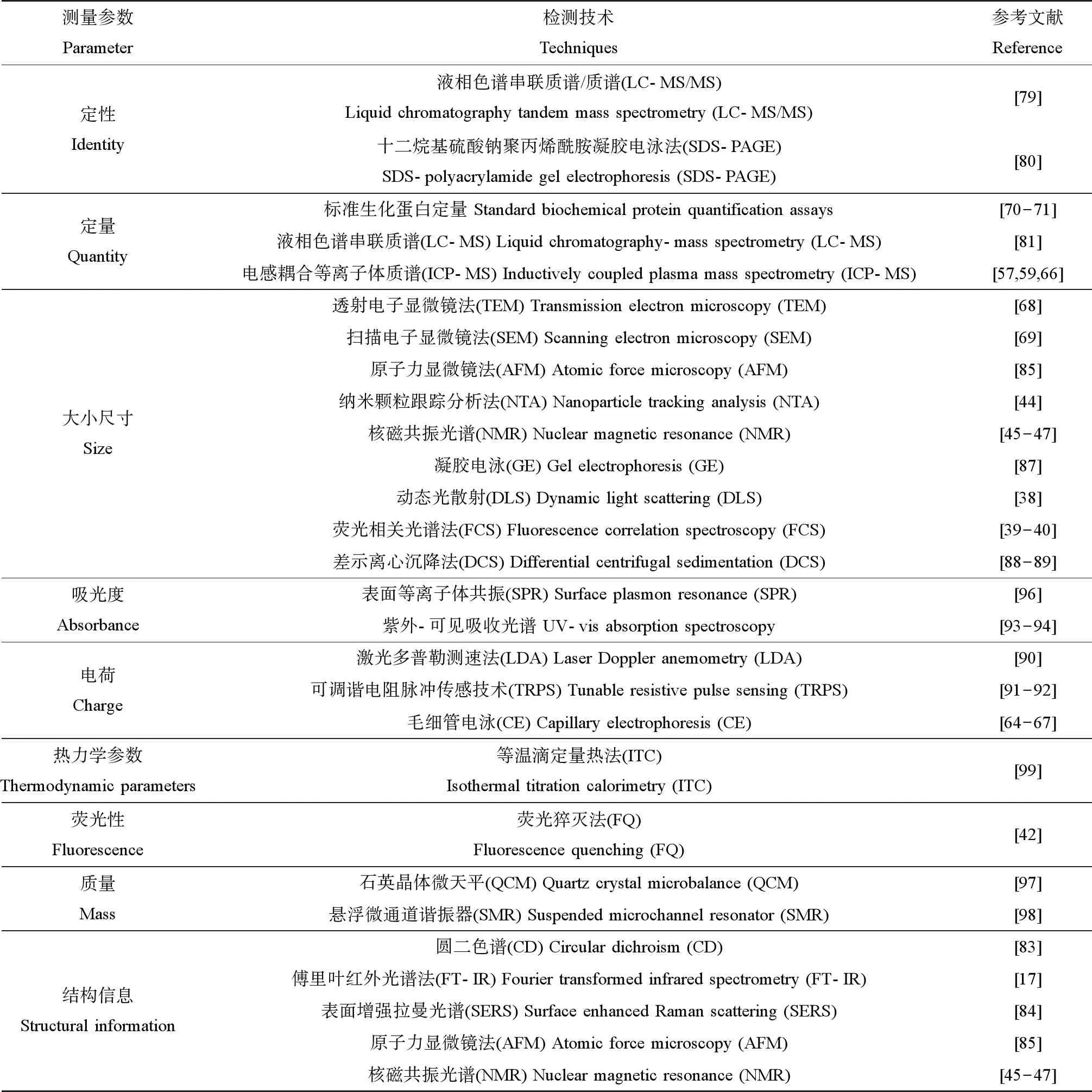
表1 蛋白冠的结构和组成表征技术Table 1 Techniques to examine the structure and composition of protein corona
2.3 蛋白冠的指纹特征
指纹特征通常是指借助一定的分析手段,得到可以标记蛋白冠组成特征的指标,以显示不同蛋白冠之间的差异,从而区分不同蛋白冠。通过对指纹特征的数据进行提取和分析,构建数学模型,获得其隐含的潜在信息。目前,用于指纹特征研究的数据处理方法主要有聚类分析法、主成分分析法和相似度分析法等。
蛋白冠分子指纹的精确鉴定一般使用MS和SERS等非靶向分析技术,如通过化学和酶处理形成肽片段,然后使用SDS-PAGE、二维色谱或蛋白质电泳联用MS等对蛋白质进行分离和鉴定。MS能够进行无标记检测,具有高通量和高灵敏度等优点。已有研究大多关注聚乙二醇化多壁碳纳米管上蛋白冠的蛋白质指纹图谱,基本都使用天然和变性缓冲液在不同条件下依次洗脱血浆蛋白,再采用SDS-PAGE、2D-PAGE分离和MS对蛋白冠进行蛋白质组学指纹图谱分析,分析鉴定出软冠中参与免疫反应调节的蛋白组分,以及硬冠中维持宿主稳态的作用生物分子[100-101]。有研究利用SDS-PAGE和MS对磁赤铁矿纳米颗粒(surface active maghemite nanoparticles,SAMNs)表面选择性吸附形成的蛋白冠进行分析和定量,发现SAMNs能够选择性吸附FALPQYLK序列的alpha(s2)-酪蛋白片段,该片段是casocidin-1肽的一部分,具有很强的抗菌活性[102]。
近期有研究者针对Gd@C82(OH)22纳米颗粒分别在健康人血浆和肺癌源性血浆孵育后形成的蛋白冠进行特征分析,发现健康人体血浆和癌症病人血浆中形成的蛋白冠在蛋白质组成和数量上都存在显著差异,表现出不同的蛋白质组指纹特征,表明蛋白冠指纹在医学诊断方面存在潜在的应用前景[103]。此外,利用三维SERS基底检测生物和医学样品的生物分析是近年来的热点[104]。有研究使用SERS得到多孔纳米结构与特定的N-乙酰-β-D-氨基葡萄糖苷酶(N-acetyl-β-D-glucosaminidase,NAG)产物的相互作用后形成的指纹图谱[105]。多孔Ag-Si材料SERS底物在复杂基质中的灵敏度即使与其他先进检测技术相比也具有足够的优势(在尿、牛奶和血浆中分别为0.07、0.47和0.50 mU·mL-1),显示了基于SERS分析的蛋白冠指纹特征在病理早期诊断方面的应用潜能。显然,对蛋白冠指纹特征的深入理解和知识储备可以为纳米颗粒的设计开辟一种新的方法,即新型纳米材料的创造性生产可以从识别蛋白冠的基本指纹模式和构象开始,以实现预期的生物和医学应用。
3 蛋白冠的生物效应(Bioeffect of protein corona)
蛋白冠的形成降低了纳米颗粒的表面能,赋予了纳米颗粒新的生物特性。蛋白冠在纳米颗粒的细胞摄入和生物分布过程中发挥重要作用,能影响纳米颗粒的生物有效性、相容性和毒性[106-107]。
3.1 减缓纳米颗粒的生物毒性
迄今,已有许多研究证实蛋白冠的包覆能减缓纳米颗粒的生物毒性。比如,氧化石墨烯在1%胎牛血清中孵育时,其对人体细胞具有明显的毒害作用,并表现出浓度依赖性的细胞毒性;但当氧化石墨烯在10%胎牛血清中孵育时,其对人体细胞的毒性显著降低[108]。机理研究证实氧化石墨烯的细胞毒性主要源于细胞膜和氧化石墨烯纳米片之间的直接相互作用,导致细胞膜的物理损伤。当氧化石墨烯在较高浓度的胎牛血清中孵育时,由于氧化石墨烯具有极高的蛋白质吸附能力,使得其表面形成蛋白冠,阻断了氧化石墨烯与细胞膜的直接接触(图2),从而降低对细胞膜的损伤。可见,蛋白冠能够通过阻断纳米颗粒与细胞膜的接触或渗透,得以减缓纳米颗粒的生物毒性[109]。
此外,研究表明蛋白冠也可以降低细胞内活性氧(reactive oxygen species,ROS)的产生(图2)。研究人员利用食源性纳米颗粒(food-borne nanoparticles,FNPs)作为模式纳米颗粒,通过人血清白蛋白孵育使纳米颗粒表面形成蛋白冠,发现蛋白冠缓解了FNPs诱导的HepG-2和正常大鼠肾(normal rat kidney,NRK)细胞中谷胱甘肽、超氧化物歧化酶、丙二醛和过氧化氢酶等氧化应激,证实了蛋白冠能降低FNPs引起的HepG-2和NRK细胞的线粒体膜电位和ROS的产生量,进而减缓FNPs的细胞毒性[110]。蛋白冠还可以通过减少细胞对纳米颗粒的吸收来减缓细胞毒性。比如,银纳米颗粒吸附血清蛋白形成蛋白冠,可显著降低小鼠胚胎成纤维细胞对银纳米颗粒的摄入,减缓其细胞毒性[111]。
3.2 增强纳米颗粒的生物毒性
研究表明,单壁碳纳米管可引发细胞增殖率降低、加快细胞衰老和死亡等毒性作用,当单壁碳纳米管表面形成硬蛋白冠后,其长期毒性增强[112]。进一步机理研究显示,碳纳米管表面形成的硬冠包含调节蛋白,能够抑制细胞的增殖。也有研究表明,在磁场作用下超顺磁铁氧化物纳米颗粒(superparamagnetic iron oxides,SPIOs)表面形成的蛋白冠会导致细胞摄入更大量的纳米颗粒,引起细胞毒性增强[113]。SPIOs的细胞毒性机制一般与摄取量和ROS含量有关[114],研究显示蛋白冠中载体蛋白含量的增加导致细胞对SPIOs摄取量的增加,进而引起细胞内ROS含量增加(图2)。然而,当猝灭细胞体系中的ROS后,细胞活性并没有显著提高,表明ROS水平的增加与观察到的细胞活性降低并无直接关系,可能还存在其他由SPIOs摄入量增加导致细胞毒性增强的替代机制。综上,蛋白冠的“双刃剑”生物效应的示例及潜在作用机制示意图如图2所示。
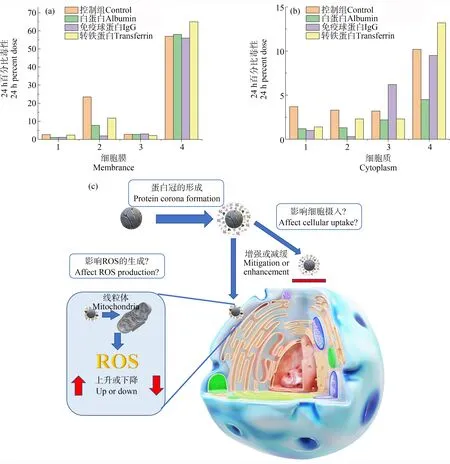
图2 蛋白冠的“双刃剑”生物效应注:(a)和(b)分别代表蛋白冠形成前后不同纳米颗粒对于人表皮角质形成细胞的细胞膜和细胞质毒性,数据总结自Monteiro-Riviere等[115]的工作;其中1、2、3和4代表不同类型纳米颗粒,1代表20 nm柠檬酸银,2代表110 nm柠檬酸银,3代表20 nm二氧化硅修饰的银颗粒,4代表120 nm二氧化硅修饰的银颗粒;(c) 蛋白冠影响纳米颗粒引起的细胞毒性强弱的机制[110]。Fig.2 The “double-edged sword”effect of protein coronasNote:(a) and (b) respectively represent the membranal and cytoplasmic toxicity of different nanoparticles to human epidermal keratinocyte before and after the formation of protein coronas;the data are collected from previous study[115];1,2,3 and 4 represent different types of nanoparticles,1 represents 20 nm citrate silver,2 represents 110 nm citrate silver,3 represents 20 nm silica-coated silver,and 4 represents 120 nm silica-coated silver;(c) mechanism of protein coronas affecting the cytotoxicity induced by nanoparticles[110].
3.3 蛋白冠的潜在医药应用
纳米给药系统在疾病靶向治疗方面具有强大的优势。与传统药物相比,纳米给药系统可增加药物的溶解度和生物相容性,减少游离药物的脱靶副作用。然而纳米给药系统仍存在一些缺点,包括纳米颗粒的低效率传递和潜在的长期毒性,这些都制约了其在临床的应用。近些年研究发现蛋白冠的持久性和动力学特性可能具有医药应用价值[116]。比如,内吞体和溶酶体的酸性和酶促环境可能在体内外调节蛋白冠,通过合成对pH和温度敏感的“智能”聚合物或包含隐形部分的共聚物或具有多层聚合物的纳米颗粒,可能有利于控制药物释放[117-118]。蛋白冠也可以通过自组装来装载药物[119]。比如,通过改变生理微环境的局部pH值和离子强度可以使聚合物具有特定的刚性,而线状或拉丝状聚合物与球状蛋白质之间的相互作用也可能会影响结构,从而影响蛋白冠的物理化学性质。这些性质的动态调节有助于调控蛋白冠修饰的纳米给药系统,减少靶向药物的受损,以实现生物体内靶向药物在到达目标之前的稳定性以及后续的药物缓释。因此,开发新型纳米颗粒结构,吸附特定的内源性生物分子到纳米颗粒表面形成蛋白冠,从而提高靶向性和药物传递仍然将是医药应用领域的重要研究课题。
4 结论及展望(Summary and perspective)
综上所述,本文重点对蛋白冠的形成机理、蛋白冠组成及结构的分析表征和蛋白冠的生物效应进行了归纳总结。目前,针对蛋白冠的研究非常宽泛,从软-硬蛋白冠的分析,到纳米颗粒物理化学特性对蛋白冠结构组成的影响,从蛋白冠影响纳米颗粒生物效应的分子机制,到蛋白冠纳米靶向载药体系的设计与应用,都显示了蛋白冠对纳米颗粒归趋和功能的重要作用。尽管在以上方面取得了一些进展,但对蛋白冠与颗粒物介导的生理疾病之间是否存在潜在的关系尚不清楚,尤其是纳米颗粒急性毒性阶段的蛋白冠组成动态变化与代谢通路相关蛋白的瞬时失稳是否存在相互关系。如果无法填补这些知识空缺,就很难深入地了解纳米颗粒的健康风险,以及创新性地设计出新型纳米靶向载药系统以实现精准医疗。此外,基于现有研究数据,建立健全相对完善的蛋白冠指纹特征数据库和模型,尝试开展更多与蛋白代谢相关的代谢冠研究,预测纳米颗粒的生物学效应,并尝试服务于临床病例诊断。
猜你喜欢
军事文摘(2022年12期)2022-07-13
肝博士(2022年3期)2022-06-30
少儿科技(2022年2期)2022-03-05
陶瓷学报(2021年4期)2021-10-14
海外星云(2021年9期)2021-10-14
北京航空航天大学学报(2017年2期)2017-11-24
中国民族民间医药·下半月(2014年4期)2014-09-26
学苑创造·A版(2009年6期)2009-12-07
环球时报(2009-09-11)2009-09-11
