
快速溶剂萃取-气相色谱法测定土壤中石油烃(C10~C40)方法研究
2022-09-27 07:20:12岳永丽
甘肃科技 2022年14期
岳永丽,付 宁
(甘肃省环境监测中心站,甘肃 兰州 730020)
石油烃是环境中普遍存在的有机污染物之一,包括汽油、柴油、煤油、润滑油、石蜡和沥青等,由多种烃类(正烷烃、支链烷烃、环烷烃、芳烃)和少量其他有机物组成[1-3]。与石油烃污染水环境不同,石油烃污染土壤环境后修复治理难度更大,修复成本也更高,《土壤环境质量 建设用地土壤污染风险管控标准(试行)》(GB 36600—2018)中针对石油烃污染物规定了相应的筛选值和管制值[4-8]。
目前,土壤中石油烃的测定方法主要有荧光光度法、紫外分光光度法、非分散红外光度法、红外分光光度法和气相色谱法等[9-12]。其中荧光光度法和紫外分光光度法只能测定石油烃中的芳烃成分;非分散红外光度法只能测定石油烃中的甲基和亚甲基,不能测定石油烃中芳烃成分[13];红外分光光度法测定石油烃的范围广,不受油品限制,但是测定过程仍有一定的局限性。一是不能反映出石油烃的组分信息,容易出现假阳性干扰测定结果;二是样品前处理过程中用到的萃取溶剂只能选择臭氧层破坏物质(ODS)或类ODS物质(四氯化碳、四氯乙烯等),ODS 已被联合国环境规划署列入加速淘汰计划[14]。相比而言气相色谱法可以采集到石油烃的指纹特征,选择性好且测定结果精确度高[15]。王如刚等[16]利用气相色谱法测定土壤中石油烃,以加标回收率为判断依据,对比了超声波提取、索氏提取及超声波-索氏提取结合3种萃取方法的萃取效率,发现超声-索氏提取结合的提取效率最佳,但在实际分析检测工作中索氏提取耗时较长,并不能及时有效地完成应急监测等工作。Tor等[17]验证了超声波萃取法-气相色谱法测定土壤中石油烃具有效率高,分析快等特点;周瑞娟等[18]通过实验验证超声波清洗仪与往复振荡器结合能够有效提取土壤中石油烃。
气相色谱法测定土壤中石油烃的研究中,不同前处理方法对测定结果影响很大,同一前处理方式中不同的萃取条件也影响测定结果。现有研究中虽然对不同前处理方式进行了比较,但是缺乏系统详细的前处理方法比较研究及条件优化,同时缺乏连续测定过程中数据结果准确度的质量控制研究,本研究比较了4种不同前处理方式索氏提取、超声波提取、震荡提取和快速溶剂萃取法(ASE)对气相色谱法测定土壤中石油烃结果的影响,对快速溶剂萃取法提取土壤中石油烃的萃取条件进行了优化,同时采用质量控制图对气相色谱法测定土壤中石油烃的加标回收率进行了统计分析,通过检查数据质量的准确度,观察实验数据的波动,从而确保分析结果准确性和可靠性,对于土壤中石油烃分析测定具有一定的借鉴作用。
1 实验
1.1 仪器和试剂
气相色谱仪(7890A,美国Agilent,氢火焰检测器FID);HP-5毛细管色谱柱(Agilent30 m×0.32 mm×0.25μm);自动索氏提取仪(SoxtecTM 8000,丹麦FOSS);ASE(ASE350,美国赛默飞);数控超声波仪(KH-500DE型);震荡器(HY-2A);自动氮吹浓缩装置(ATR AutoVapⅡ,美国ATR)。
石油烃(C10~C40)正构烷烃标准溶液(每种烷烃的质量浓度分别为1000 mg/L,溶剂为正己烷);土壤中石油烃(C10~C40)有证标准物质(总石油烃浓度为1100 mg/kg,美国ERA公司);二氯甲烷、正己烷、丙酮、石油醚(均为色谱纯,J.T.BAKER,USA);硅酸镁净化柱(美国Waters);玻璃棉(马弗炉中500 ℃下烘烤4 h);无水硫酸钠(于马弗炉中500 ℃下烘烤4 h);高纯氮气(纯度>99.999%)。
1.2 色谱条件
气化室温度:320 ℃,分流比5∶1;
柱温箱程序升温:60 ℃保持4 min,以10 ℃/min升至230 ℃保持2 min,再以20 ℃/min升至230 ℃保持20 min;
FID燃气流量为400 mL/min,助燃气流量为30 mL/min,尾吹气流量为30 mL/min;柱流失控制为恒流模式,载气流速为1.0 mL/min。
检测器温度:330 ℃。
1.3 实验步骤
1.3.1 土壤样品土壤样品TR-1、TR-2、TR-3取自甘肃省平凉市泾川县柴油罐车泄漏后附近土样。
1.3.2 样品制备
将新鲜土壤除去样品中的异物(石子、树枝)等,称取两份10.0 g新鲜土壤样品,一份用来测定含水率及干物质含量,另一份用于土壤中石油烃的提取。
1.3.3 样品前处理
1)样品提取。索氏提取法:将样品放入提取器中,加入40 mL正己烷/丙酮溶液(V/V=1),90 ℃水浴,提取16~18 h后,转移氮吹浓缩至3~5 mL待净化。
超声波提取法:将样品放入250 mL三角瓶中,加入40 mL正己烷/丙酮溶液(V/V=1),超声频率为40 kHz,超声30 min,超声提取两次合并有机相,后转移氮吹浓缩至3~5 mL待净化。
震荡提取法:将样品放入250 mL三角瓶中加40 mL正己烷/丙酮溶液(V/V=1),以200次/min往复振荡30 min,振荡提取两次合并有机相,后转移氮吹浓缩至3~5 mL待净化。
ASE萃取法:将样品与硅藻土搅拌均匀后放入萃取池(33 mL)中,设置萃取温度、静态萃取次数及萃取时间后,利用有机溶剂萃取,萃取液浓缩至3~5 mL待净化。ASE萃取优化条件实验中利用正交实验系统分析萃取有机溶剂,萃取温度、静态萃取次数及萃取时间对萃取效率的影响。
2)样品净化。利用硅酸镁柱净化,依次用10 mL正己烷/二氯甲烷溶液(V/V=4)的混合溶剂、10 mL正己烷活化净化小柱,待正己烷近干时,将提取浓缩液转移至硅酸镁柱上,用2 mL的正己烷洗涤氮吹管,再用10 mL正己烷/二氯甲烷溶液(V/V=4∶1)的混合溶剂淋洗,收集洗脱液并氮吹浓缩定容至1 mL上机。
2 结果与讨论
2.1 石油烃(C10~C40)标准色谱图
气相色谱法测定石油烃(C10~C40)是以C10~C40出峰时间段内出峰的总面积进行定量分析。程序升温会引起柱流失而导致色谱峰基线升高,影响色谱分析定量结果,因此,需要扣除柱流失。图1(a)和图1(b)分别为石油烃标准溶液(1550 mg/kg)扣除柱流失前、后的色谱图,结果显示扣除柱流失后色谱图基线明显降低,色谱峰的响应减小,色谱峰总面积减小。
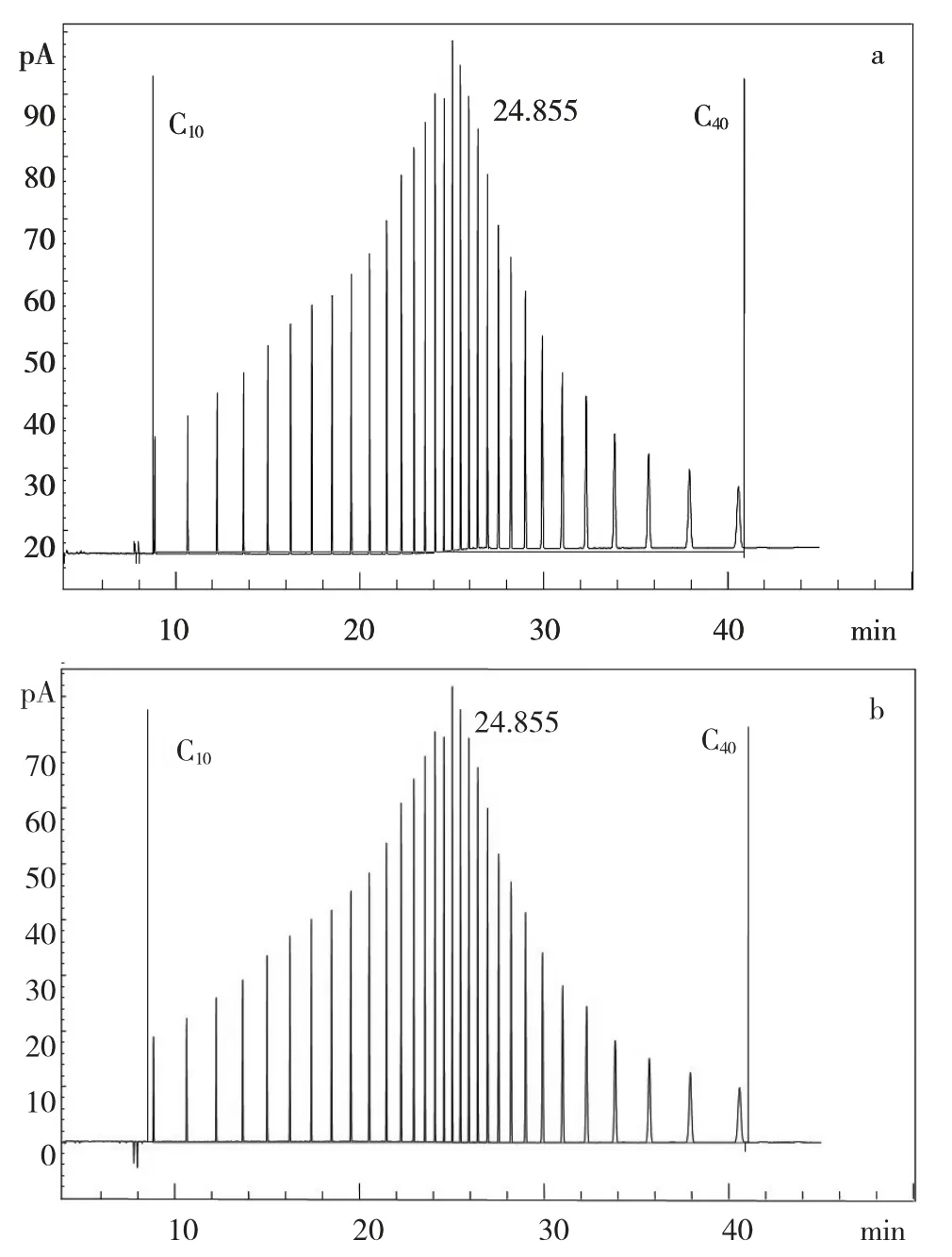
图1 石油烃(C10~C40)标准溶液扣除柱流失前(a)及扣除柱流失后(b)的色谱图
2.2 石油烃(C10~C40)标准曲线
将石油烃(C10~C4)0正构烷烃标准溶液用正己烷溶剂配制质量浓度为155、310、620、1 550、2 480、3 100、6 200 μg/mL的石油烃(C10~C40)正构烷烃的标准曲线系列点,按照1.5的仪器工作条件进行上机分析。以石油烃(C10~C40)正构烷烃之间31个色谱峰的总面积对其质量浓度绘制外标法校准曲线,图2为扣除柱流失后的校正总峰面积与浓度绘制的外标法标准曲线图,结果表明石油烃(C10~C40)线性关系良好,能够满足分析需要。
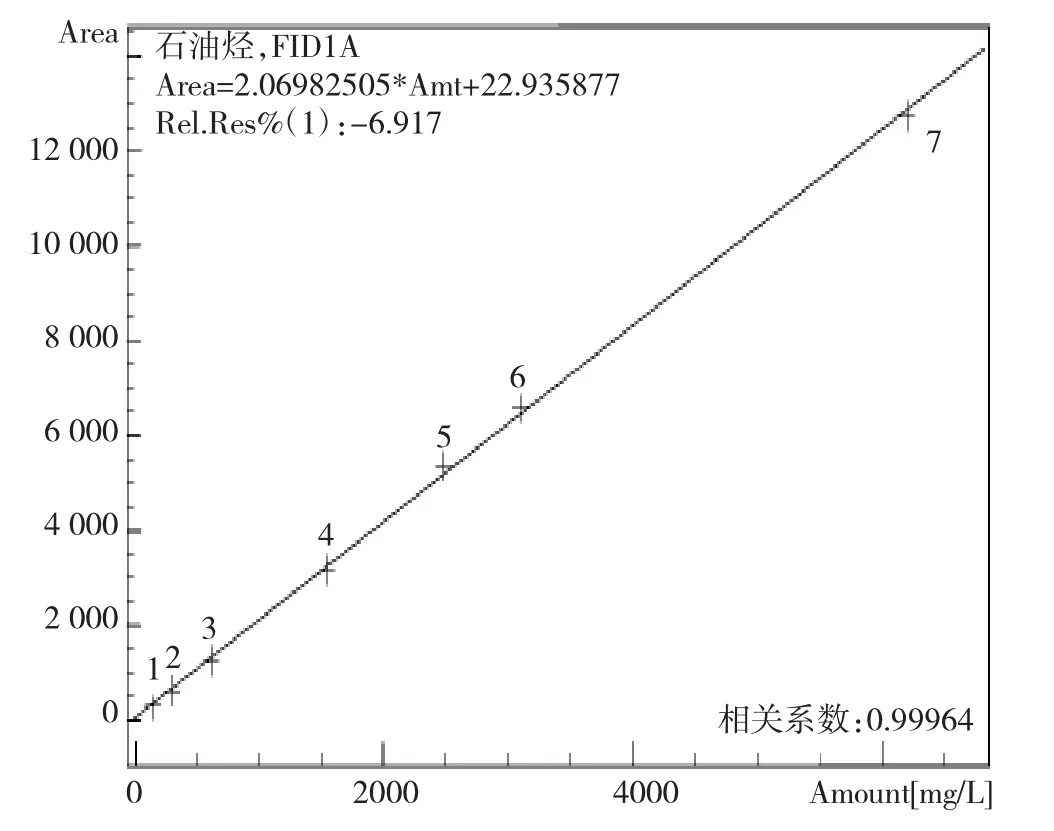
图2 石油烃(C10~C40)线性方程及相关系数
2.3 实验条件的优化
2.3.1 提取方法
准确称取10.0 g石英砂,加入20 μL石油烃(C10~C40)标准溶液(加标浓度为62 mg/kg),40 mL正己烷与丙酮(V/V=1)为萃取剂,分别利用快速溶剂萃取、索氏提取、超声波提取和水平震荡提取4种萃取方式平行测定6次,测定加标回收率,结果表明(见表1),快速溶剂萃取和索氏提取的平均回收率分别为100%和99.0%,超声提取和水平震荡提取的平均回收率分别为85.6%及78.0%,ASE和索氏提取的萃取效率更高,但是鉴于快速溶剂萃取具有萃取溶剂消耗量少、萃取时间短、萃取速度快、萃取效率高、自动化程度高等优势,ASE更适合萃取土壤中石油烃。
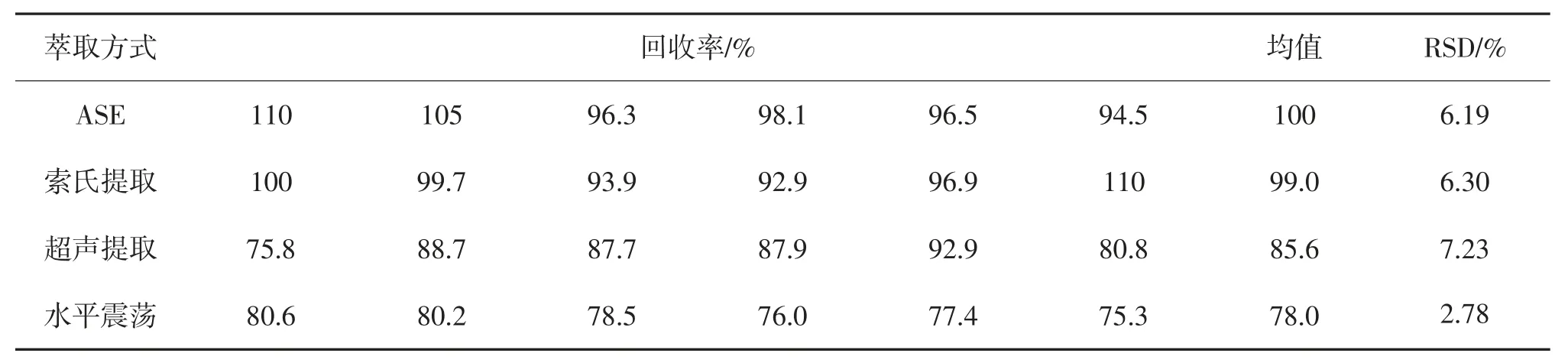
表1 不同萃取方式提取效率比较
2.3.2 ASE萃取条件优化
影响ASE萃取效率的主要条件有萃取溶剂、萃取温度、静态萃取次数及萃取时间,因此,为了进一步研究ASE萃取条件对萃取效率的影响,选择正己烷与丙酮(V/V=1)的混合溶剂、二氯甲烷: 丙酮的混合溶剂(V/V=1)、石油醚三种不同萃取溶剂,设计四因素三水平正交试验,见表2。根据正交试验设计法对萃取溶剂、萃取温度、循环次数以及静态萃取时间同时进行优化,根据极差分析确定影响因素的主次顺序,最终确定最优条件的组合。表3中正交试验设计的极差分析结果可知,影响萃取效率因素的主次顺序依次为:萃取温度(B)>萃取试剂(A)>静态萃取时间(C)>循环次数(D)。在设定的水平范围内,萃取试剂与萃取温度项的极差最大,对萃取效率最为明显;循环次数项的极差最小,对萃取效率影响不明显。石油醚的极性较弱,萃取效果不理想,萃取效率最低;因此,正交试验表明ASE最佳萃取条件为:正己烷:丙酮(V/V=1)的混合溶剂为萃取剂,萃取温度为105 ℃,静态萃取时间为10 min,循环萃取为3次。
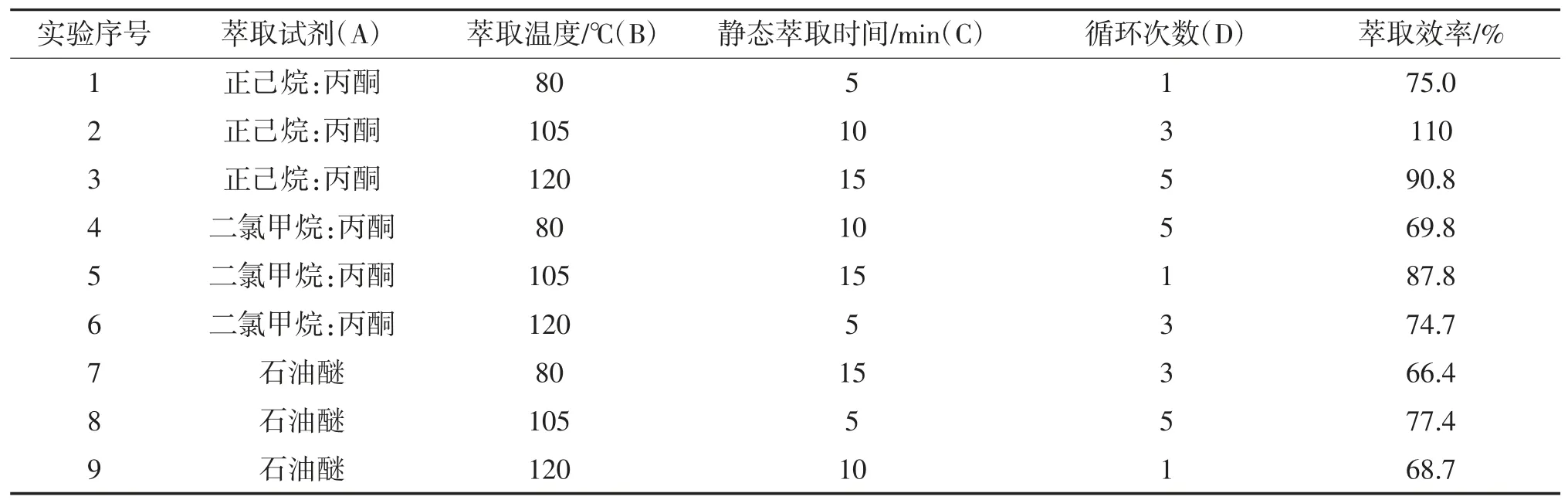
表2 正交试验设计表

表3 正交试验极差分析结果
2.4 检出限、精密度及准确度实验
准确称取10.0 g石英砂,利用优化后的ASE萃取方法,按照实验方法平行测定7份空白加标样品,根据《环境监测分析方法标准制修订技术导则》(HJ 168—2010)[19]中附录A1.1的计算公式MDL=3.143×S计算检出限,气相色谱法测定土壤中石油烃的检出限为6 mg/kg。同时利用ASE最佳萃取条件进行了精密度、准确度实验,选择石油烃低质量浓度31.0 mg/kg、中质量浓度62.0 mg/kg和高质量浓度155 mg/kg进行空白加标实验,每组各做了6个平行实验,计算相对标准偏差(RSD)和回收率;测定土壤中石油烃有证标准物质(1100 mg/kg),每组平行测定6次,计算其相对误差,精密度和准确度测定结果见表4,三个不同浓度空白加标测定结果的平均值分别为29.8 mg/kg、58.3 mg/kg和150 mg/kg,平均回收率分别为96.3%、94.0%和96.9%,RSD分别为5.24%、2.06%和5.52%。土壤有证标准物质测定结果的平均值为1138 mg/kg,相对误差为14.1%。结果表明利用ASE-GC分析土壤中石油烃具有较好的精确度和准确度。
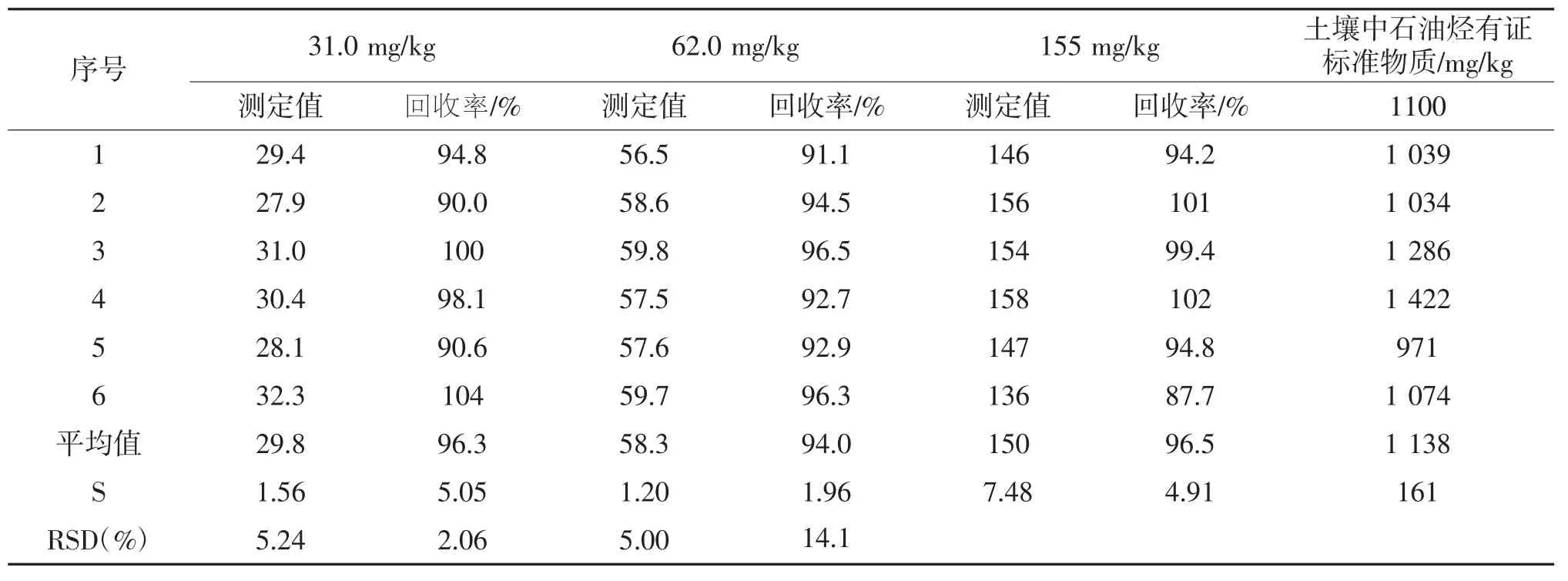
表4 精密度和准确度测定结果
2.5 实际样品分析
分别对实际土壤样品TR-1,TR-2,TR-3进行分析,并进行样品加标的测定(加标浓度为62 mg/kg),测定结果见表5,结果显示土壤样品TR-1,TR-2,TR-3平行样测定结果相对偏差均<20%,且样品加标回收率均>80.0%。图4为TR-3实际样品及加标样品色谱图,由于TR-3样品为石油泄漏后污染土壤,实际样品明显具有特征石油烃柴油色谱峰,加标样品谱图中具有实际样品柴油特征色谱峰,同时具有石油烃(C10~C40)特征色谱峰,峰面积明显增加,因此该分析方法可以准确有效测定污染土壤中石油烃。

表5 实际样品分析结果

图4 土壤TR-3实际样品(a)及加标样品(b)色谱图
2.6 质量控制图分析
环境监测分析过程中,质量控制图可以有效检查分析过程的准确性,确保分析数据准确可控[20-23]。通过对连续40个批次的实际样品加标回收率统计,计算40组样品加标回收率的算术平均值(X)和标准偏差(S),上、下控制限(X±3S),上、下警戒限(X±2S),上、下辅助限(X±S)(见表6)并绘制质量控制图(图5)。结果显示土壤中石油烃的受控范围为65.9%~119%,警戒限为74.6%~110%,辅助限为83.5%~101%。质量控制图结果表明,所有数据点都落在上下警戒限范围内;数据结果均在中心线两侧随机排列,上、下辅助限内有29个数据点,占统计点数的72.5%,大于68.3%,符合正态分布概率;没有出现连续6个点出现在中心线(平均值)的同一侧,也没有连续6个点出现递增或递减趋势。质量控制图统计分析结果表明实际样品分析过程处于统计控制状态,分析结果准确可靠[24]。

图5 土壤中石油烃分析数据质量控制图
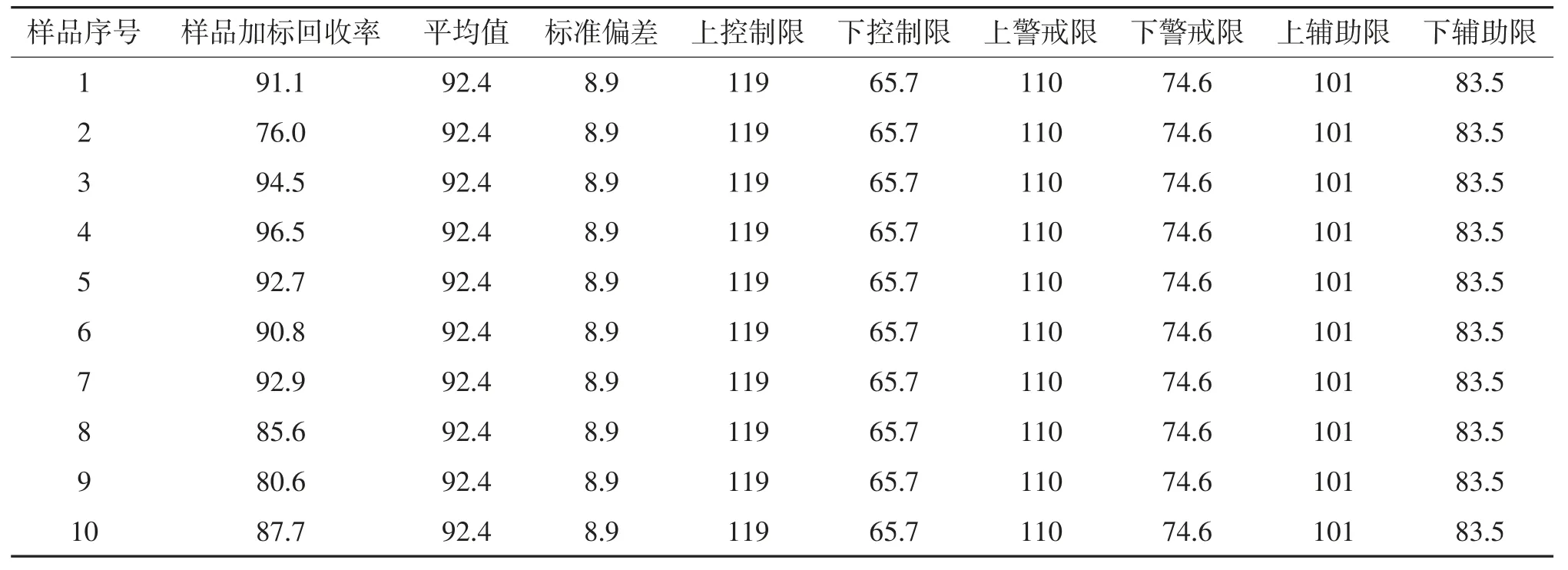
表6 土壤中石油烃(C10~C40)样品加标回收率质量控制图数据/%
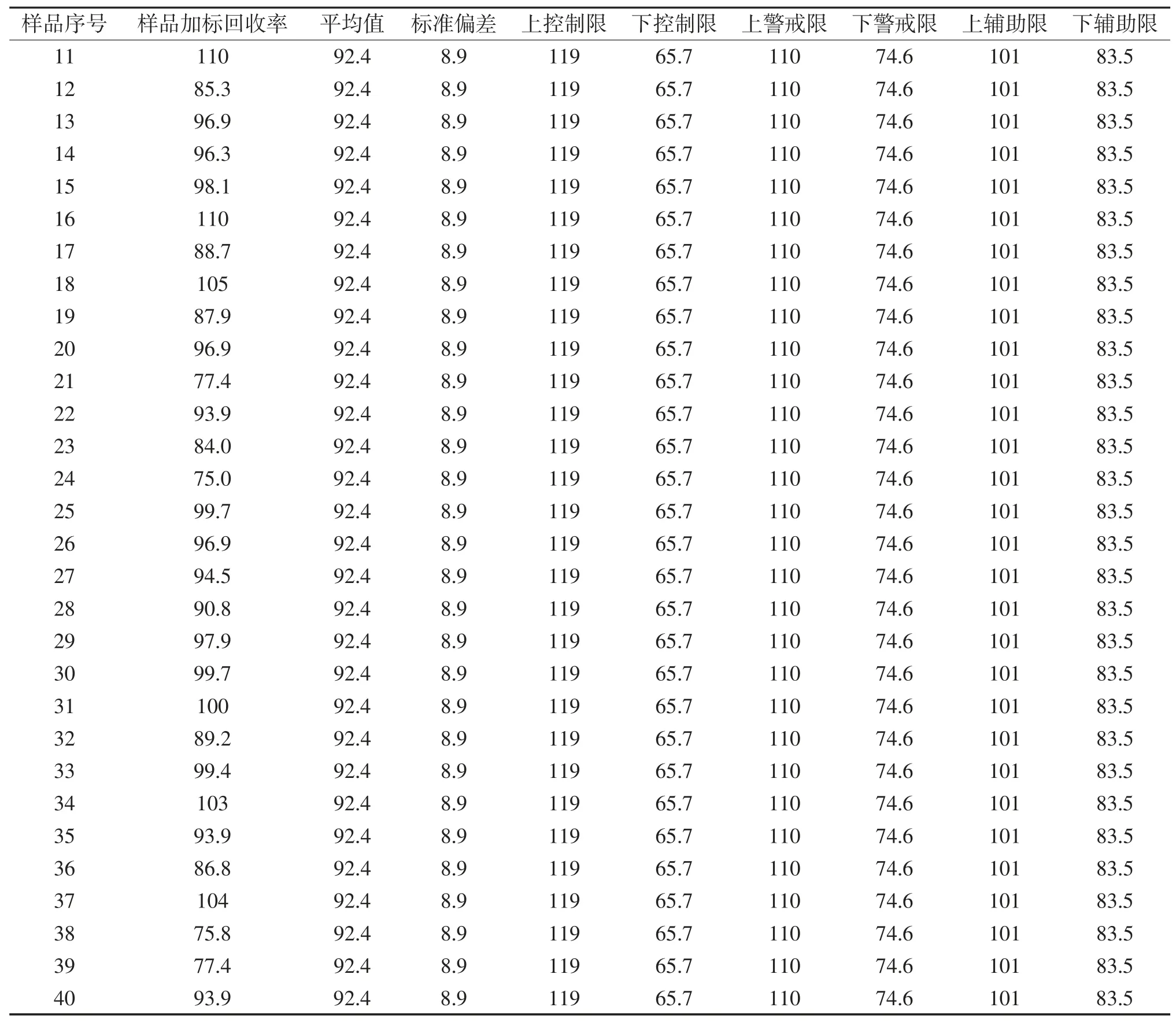
续表6
3 结论
(1)通过比较不同萃取方法并优化萃取条件,建立了有效的ASE-GC/FID测定土壤中石油烃(C10~C40)的分析方法。
(2)在最优快速溶剂萃取条件下,气相色谱法测定土壤中石油烃的检出限为6 mg/kg,该方法准确度、精密度良好,实际样品测定平行样的相对偏差<20%,且加标回收率均>80%,定量准确,能够满足土壤中石油烃(C10~C4)0的分析要求。
(3)实际分析过程中的质量控制图结果表明土壤中石油烃实际样品加标回收率的受控范围为65.9%~119%,警戒限为74.6%~110%,辅助限为83.5%~101%,所有数据点都落在上下警戒限范围内;数据结果均在中心线两侧随机排列,上、下辅助限内有29个数据点,占统计点数的72.5%,大于68.3%,符合正态分布概率。没有出现连续6个点出现在中心线(平均值)的同一侧,也没有连续6个点出现递增或递减趋势,说明分析过程处于统计控制状态,能有效地控制该方法测定土壤中石油烃(C10~C40)测定结果的准确性。
猜你喜欢
石油炼制与化工(2022年2期)2022-02-15 11:42:26
环境保护与循环经济(2021年7期)2021-11-02 08:10:52
食品安全导刊(2021年20期)2021-08-30 06:39:22
应用化工(2021年4期)2021-05-20 09:43:36
原子与分子物理学报(2021年1期)2021-03-29 07:28:54
化工管理(2020年26期)2020-10-09 10:05:16
山东化工(2019年2期)2019-02-21 09:29:32
浙江大学学报(工学版)(2016年11期)2016-06-05 09:21:04
合成技术及应用(2015年2期)2016-01-10 10:30:13
装备环境工程(2015年4期)2015-02-28 01:20:06
