
电感耦合等离子质谱法测定蟹黄中4种重金属含量的不确定度评定
2022-09-27 12:05:54姚成虎杨启鹏洪成山王岁岁毛小庆
现代农业科技 2022年18期
姚成虎 王 滢 杨启鹏 洪成山 王岁岁 孙 涓 毛小庆
(马鞍山市产品质量监督检验所,安徽马鞍山 243000)
随着工业迅速发展,我国经济水平不断提升,与此同时也给环境造成了一定影响。水产动物的饮食和水质等受到污染,导致水产动物摄入和沉积重金属的概率大大增加。重金属被人体吸收后,对造血系统、肾脏、生殖功能等器官造成不同程度的损伤[1-5]。目前,重金属检测方法主要包括紫外分光光度法[6]、原子吸收光谱法[7-9]、原子荧光光谱法[10-11]和电感耦合等离子质谱法[12-14],其中ICP-MS法凭借其高通量、快速、灵敏度高等优点成为分析痕量重金属含量的重要手段[15]。
在分析检测过程中,样品前处理、标准样品配制、标准曲线拟合、随机误差和系统误差等影响试验数据的准确性,尤其数值处于限量值的边界时,应在检测报告中进行结果的不确定度评价。因此,本文根据相关标准和规定[16-19],结合2021年度国家食品安全监督抽检任务,采用ICP-MS法对蟹黄中铅、砷、镉和铬4种重金属的不确定度来源进行了分析和评估,找出检测过程中影响测量值准确性的主要因素,以期为评定蟹黄样品中铅、砷、镉和铬4种重金属的不确定度提供参考。
1 材料与方法
1.1 材料与仪器
1.1.1 供试材料。试验样品为蟹黄,市场随机抽检。
1.1.2 试剂。供试试剂有内标溶液(含10 μg/mL的Bi、Ge、In、Li、Sc、Tb 和 Y,美国 PE 公司)、多元素混标(2.5 μg/mL,钢研纳克检测技术股份有限公司)、硝酸(UP级,68%,江苏晶瑞化学股份有限公司)、Milli-Q制备一级水。
1.1.3 仪器与设备。供试仪器有NexION 350X电感耦合等离子体发射光谱-质谱联用仪(含形态分析)(美国PE公司)、BSA124S电子天平(赛多利斯科学仪器(北京)有限公司)、MARS6微波消解仪(美国CEM公司)、EHD-24赶酸仪(北京东航科仪仪器有限公司)。
1.2 试验方法
1.2.1 标准曲线配制。吸取2.5 μg/mL混标溶液2 mL于50 mL容量瓶中,用1%HNO3稀释至刻度,得到浓度为100 μg/L的混标溶液。分别吸取0.50、2.50、5.00、15.00、25.00 mL的混标溶液置于50 mL容量瓶中,用 1%HNO3稀释至刻度,得到浓度为 1、5、10、30、50 μg/L的系列标准曲线溶液。
1.2.2 样品制备。参照《食品安全国家标准 食品中多元素的测定》(GB 5009.268—2016)中样品前处理步骤[20],准确称取6份0.25 g(精确至 0.001 g)蟹黄样品于微波消解罐中,加入5 mL硝酸,加盖放置过夜后旋紧罐盖放入微波消解仪中,按照微波消解仪操作流程进行消解。冷却后取出,于100℃条件下赶酸0.5 h,转移消解液于25 mL容量瓶中,用水定容同时做空白试验。
1.2.3 测定。电感耦合等离子质谱仪正常开机通过调试后对标准溶液进行分析,将标准溶液待测元素质谱信号与内标元素质谱信号的比值Y作为纵坐标、待测元素浓度X作为横坐标,绘制标准曲线。随后分析样品溶液,从标准曲线中得到相应浓度。ICP-MS的工作参数如下:射频功率为1 100 W,等离子体流量为15.0 L/min,雾化器气流量为0.9 L/min,辅助气流量为1.20 L/min,蠕动泵流速为20 r/min,测定次数共3次,雾化器为同心雾化器,采用KED模式。
2 结果与分析
2.1 不确定度评定的数学模型及计算公式
测量不确定度的评定方法包括A类和B类两种[16],A类评定即能采用统计学方法对在指定测定条件下获得的量值进行测量不确定度分量的评定,不同于A类评定的即为B类评定。不确定度评定公式如下:
式中:X为待测试样中重金属含量(mg/kg);C为根据标准曲线计算的待测样品中重金属含量(μg/L);C0为根据标准曲线计算出来的标准空白液中重金属含量(μg/L);V 为消化液定容体积(mL);m 为待测样品质量(g);换算系数为 1 000。
A类评定公式如下:
式中:n 为测量次数;xi为测量值(i=1,2,…,n);为测量平均值。
B类评定公式如下:
2.2 不确定度来源分析
根据测定样品的具体操作流程(图1)和数学模型分析,电感耦合等离子质谱法测定蟹黄中4种重金属含量的不确定度分量来源见表1。

表1 不确定度分量的来源及其相对标准不确定度的表示
2.3 不确定度的评定
2.3.1 样品前处理引入的相对标准不确定度urel(M)。主要包括样品称量过程和消化液定容过程引入的不确定度分量。
(1)样品称量过程引入的不确定度分量u1(M)。称量样品所用的天平引入的不确定度分量选用B类方法评定,按矩形分布。根据《电子天平检定规程》(JJG 1036—2008)[21],实验室称量待测样品所用的天平经检定最小量程为0.1 mg,分度值e为1 mg,允许最大误差为±1.0 mg,本次称量的6份样品重量均值为0.253 7 g。
(2)消化液定容过程引入的不确定度分量u2(M)。消化液定容至25 mL容量瓶,根据《常用玻璃量器检定规程》(JJG 196—2006)[22],当温度为 20 ℃时,25 mL A级容量瓶的允差为±0.03 mL,按矩形分布。
考虑到实验室温度波动范围为(20±2)℃,水的膨胀系数为 2.1×10-4(20℃),按照 95%的置信概率(k=1.96)。
2.3.2 标准溶液配制引入的相对标准不确定度urel(C)。其主要包括标准溶液的相对标准不确定度和标准物质定容过程中引入的相对标准不确定度。
(1)标准溶液的相对标准不确定度u1(C)。根据标准物质证书已知铅、砷、镉、铬的浓度为2.5 μg/mL,不确定度为U=10%(k=2),则由标准溶液引入的相对标准不确定度。
(2)标准物质定容过程中引入的相对标准不确定度 u2(C)。根据《常用玻璃量器检定规程》(JJG196—2006)[22],介绍A级玻璃量器的允差,按矩形分布。考虑到实验室温度波动范围为(20±2)℃,查得水的膨胀系数为 2.1×10-4(20℃),按照95%的置信概率(k=1.96),则由温度波动引入的不确定度分量。
在标准溶液配制过程中,配制器具单次引入的相对标准不确定度见表2。
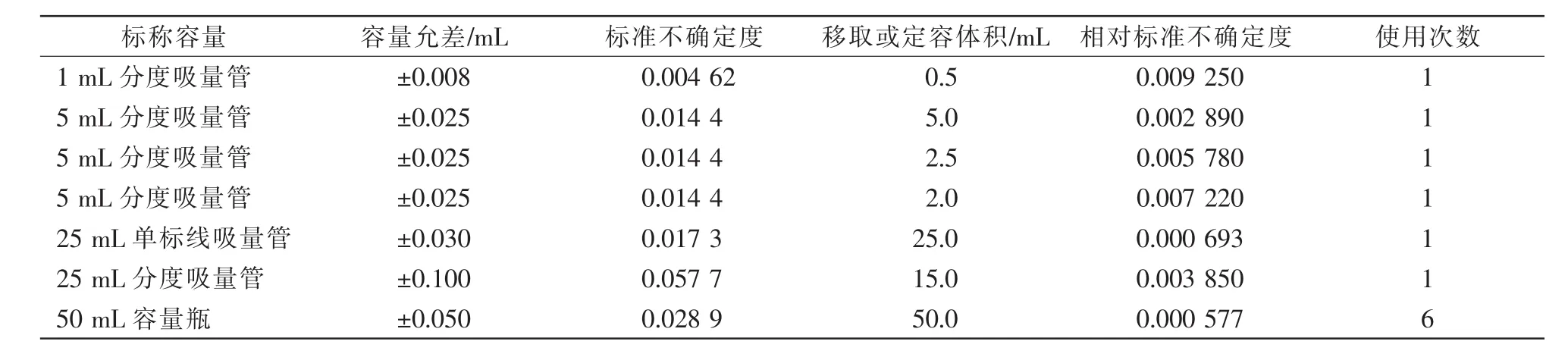
表2 配制器具引入的相对标准不确定度
考虑配制器具的使用次数,标准物质定容过程中引入的相对标准不确定度,故标准溶液配制引入的相对标准不确定度。
2.3.3 由标准曲线拟合引入的相对标准不确定度urel(X)。 分别对 5 个浓度(1、5、10、30、50 μg/mL)的标准溶液进行3次重复测定,采用内标法定量,用最小二乘法拟合得到线性方程及相关系数r,标准曲线拟合引入的相对标准不确定度 urel(X)由公式(1)和(2)进行计算。
其中:SR为残差标准偏差;a为标准曲线斜率;n为标准溶液重复测定的总次数,即n=15;xi为拟合曲线第i个标准溶液的浓度(μg/L),p为待测样品平行测量的次数,即p=3;为拟合曲线标准溶液浓度的平均值,即为待测样品测量的平均值(μg/L)。标准曲线拟合引入的相对标准不确定度urel(X)详细结果见表3。
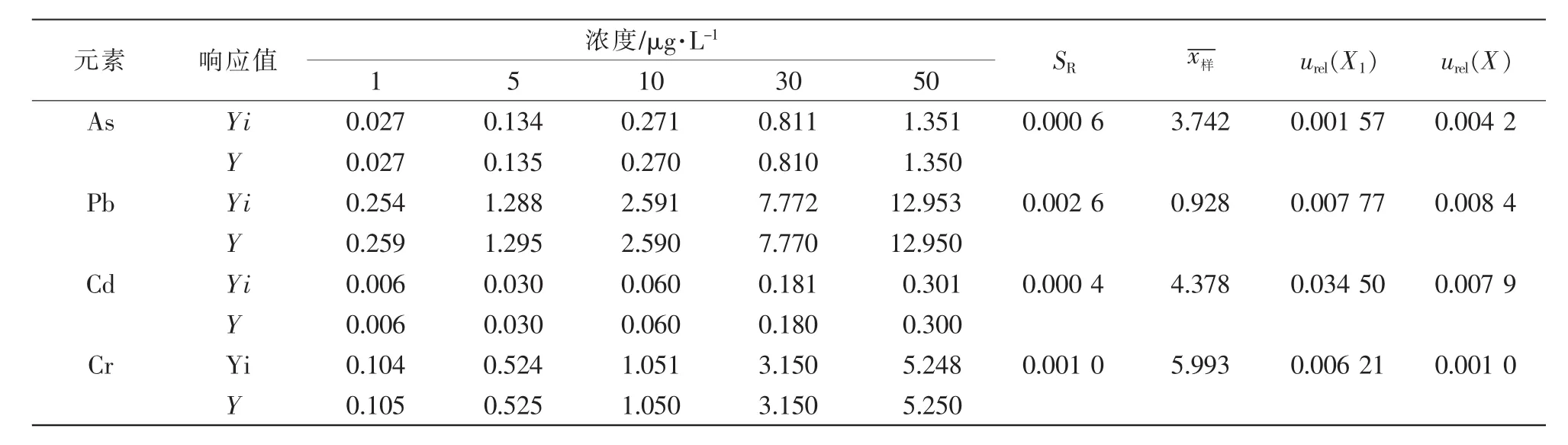
表3 标准曲线拟合引入的相对标准不确定度
2.3.4 系统误差引入的相对标准不确定度urel(Z)。在样品检测过程中,如样品的称量、消解、定容和仪器测量,可能会影响待测元素含量,日常分析中通过加标回收方式判断试验系统误差大小[23-24],再依据统计学t方法检验判断系统误差是否显著。
称取6份待测样品,加入适量体积的标准溶液,按照1.2.2进行样品前处理。根据公式(3)计算系统误差引入的相对标准不确定度urel(Z),根据公式(4)计算t。详细计算结果见表4。
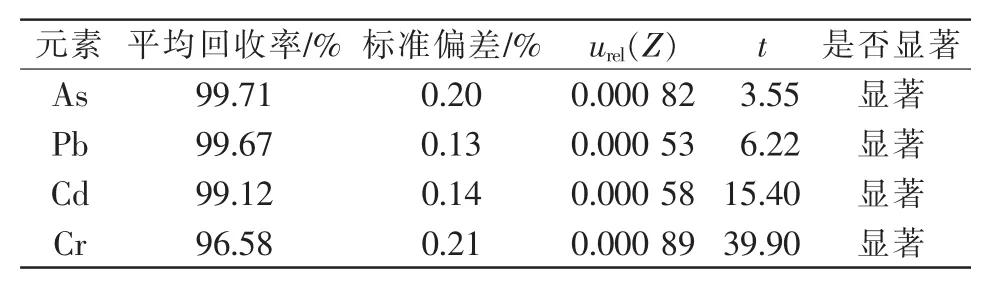
表4 系统误差引入的相对标准不确定度
其中:S为6份加标回收的相对标准偏差;s为6份加标回收的标准偏差;n为平行样品个数,n=6;为平均回收率。
取置信概率为 95%,查表得 t(0.05,5)=2.571,而此时4种元素的t值均大于2.571,故回收率与100%之间存在显著差异。因此,实际样品的检测结果需将回收率带入不确定度评定的数学模型中来修正。
2.3.5 随机误差引入的相对标准不确定度urel(S)。在试验过程中,会有众多的随机因素影响试验结果准确性,因而采用多次测量的方法来减少随机误差,根据《测量不确定度评定与表示》(JJF 1059.1—2012)[17]中相关规定,当测量次数较少时,可采用极差法评定不确定度,其中:R为极差;n为测量次数,n=6;C 为极差系数(n=6 时,C=2.53)。 详细试验结果见表5。
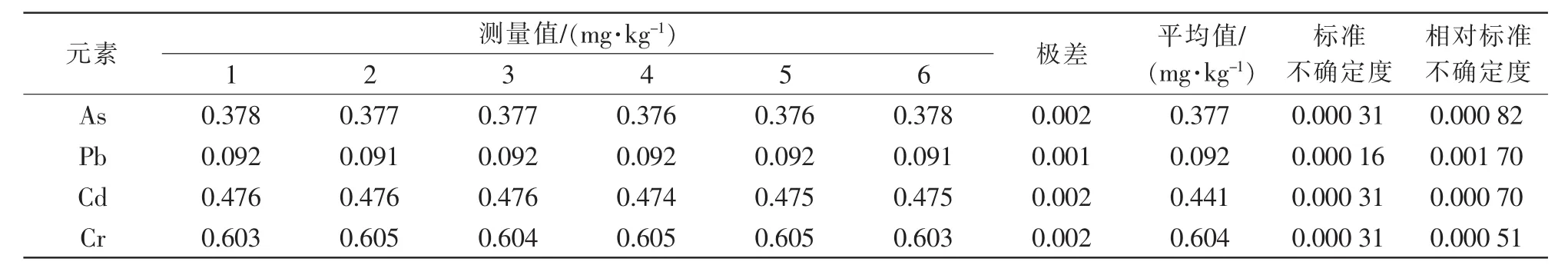
表5 随机误差引入的相对标准不确定度
2.4 合成标准不确定度
将上述步骤中所有的相对标准不确定度进行合成,合成公式按照式(5)进行计算,假设包含因子k=2,则合成后的相对标准不确定度和扩展不确定度详细结果见表6。
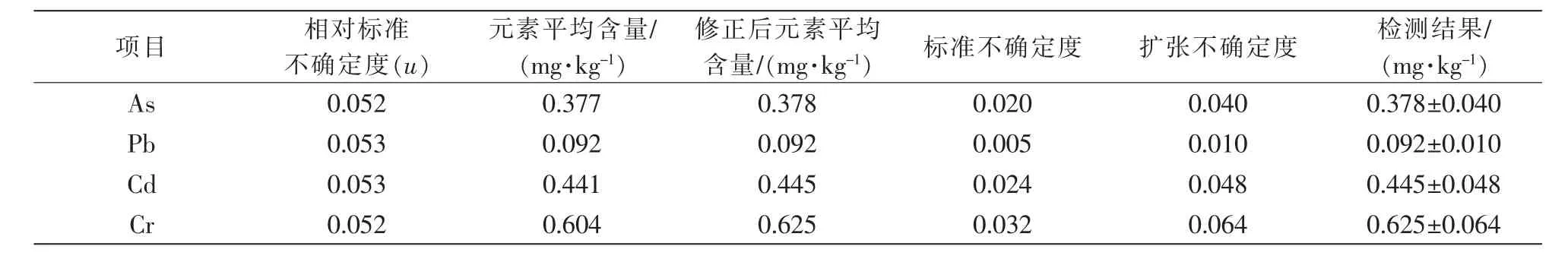
表6 各元素的标准不确定度结果
2.5 相对标准不确定度分量占比汇总
各元素相对标准不确定度分量占比汇总见图2。
3 结论与讨论
采用电感耦合等离子质谱法测定蟹黄中4种重金属含量,并对其不确定度来源进行了分析。结果显示,铅、砷、镉和铬的扩展不确定度分别为0.040、0.010、0.048、0.064 mg/kg(k=2);不确定度来源主要为标准溶液配制、样品前处理和标准曲线拟合。
在标准溶液配制过程中,稀释次数与不确定度引入的影响成正比;当测定的元素含量很低时,标准曲线拟合引入的不确定度影响较大。因此,可以通过以下途径来减少不确定度带来的影响:①减少标准溶液稀释次数;②选择合适的消解程序以及赶酸条件;③对于痕量元素的分析,标准曲线线性范围尽量窄,确保待分析元素的含量在标准曲线的中间位置;④待测溶液和标准溶液的基体保持一致;⑤使用有证书且在有效期范围内的标准物质;⑥定期对仪器进行维护保养,提高仪器灵敏度;⑦重视对检验人员理论知识的考核和试验操作规范性的培养;⑧保持实验室温湿度的恒定,尽量减少波动;⑨增大样品重复分析测定次数。
猜你喜欢
疯狂英语·初中版(2023年3期)2023-10-25 16:53:09
快乐作文(1.2年级)(2021年2期)2021-09-10 21:22:48
华人时刊(2020年17期)2020-12-14 08:12:56
价值工程(2017年31期)2018-01-17 00:34:27
电子测试(2017年12期)2017-12-18 06:35:46
水利科技与经济(2016年7期)2016-04-25 13:03:12
水利科技与经济(2016年8期)2016-04-22 03:41:22
电源技术(2015年7期)2015-08-22 08:48:52
电测与仪表(2015年7期)2015-04-09 11:40:30
小主人报(2015年2期)2015-02-28 20:42:42
