
超高效液相色谱-电雾式检测器测定福建产绞股蓝中绞股蓝皂苷XLVI和LVI
2022-09-26 07:01卢彭信梁海珍柴瑞平罗定强郭宝林马百平
色谱 2022年9期
卢彭信, 李 港, 郑 伟, 梁海珍, 张 洁,柴瑞平, 罗定强, 金 燕, 郭宝林, 马百平*
(1. 广东药科大学中药学院, 广东 广州 510060; 2. 军事科学院军事医学研究院辐射医学研究所, 北京 100850;3. 赛默飞世尔科技(中国)有限公司, 上海 201206; 4. 中国医学科学院北京协和医学院药用植物研究所, 北京 100193; 5. 陕西省食品药品检验研究院, 陕西 西安 710065)
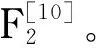
绞股蓝尚未收录至2020版《中国药典》,仅收录于地方标准,如在《福建省中药材标准(2006年)》中,对绞股蓝的质量控制仅局限于性状、色味,对绞股蓝总皂苷或具体皂苷无明确的含量控制。根据文献调研发现[17-20],目前关于绞股蓝质量控制的液相色谱法,多采用紫外(ultraviolet, UV)检测器测定绞股蓝中一种或多种皂苷的含量。然而皂苷类化合物通常无紫外吸收或仅为末端吸收,UV对于这类化合物的检测不稳定,灵敏度低,并且在梯度洗脱时容易出现基线漂移。电雾式检测器(charged aerosol detector, CAD)是一种质量相关的通用型检测器[21],对比其他通用型检测器如蒸发光散射检测器(evaporative light scattering detector, ELSD)和示差折光检测器(differential refractive index detector, RID), CAD具有灵敏度更高[22]、重现性更好和检测范围更宽[23]等特点,其检测信号不依赖于被测物质的化学结构,更适用于无紫外吸收或只有较弱紫外吸收成分的定量分析[24]。
本研究利用UHPLC-Q-TOF/MS结合UHPLC-CAD鉴定了福建产绞股蓝的主要成分,经过碱水解后[25],绞股蓝皂苷XLVI(gypenoside XLVI)和绞股蓝皂苷LVI(gypenoside LVI)含量远高于其他皂苷成分,为福建产绞股蓝中的主成分,适合作为福建产绞股蓝质量评价的指标成分,且目前缺少相关报道。因此,本研究利用UHPLC-CAD建立了福建产绞股蓝中绞股蓝皂苷XLVI和LVI的含量测定方法,为福建产绞股蓝药材的质量评价和标准制定提供理论参考。
1 实验部分
1.1 仪器、试剂与材料
Vanquish Flex UHPLC液相色谱仪、CAD检测器(美国Thermo Fisher公司); Waters ACQUITY I-Class超高效液相色谱系统、VION-IMS-Q-TOF质谱系统(美国Waters公司); BP 211D十万分之一天平(德国Sartorius公司); KQ-600DE数控超声波清洗器(昆山市超声仪器有限公司)。
乙腈(质谱纯,美国Thermo Fisher公司);蒸馏水(广州屈臣氏有限公司);无水乙醇(分析纯,国药集团化学试剂有限公司)。对照品绞股蓝皂苷XLVI和LVI由本课题组从福建产绞股蓝中分离得到,经HR-ESI-MS以及1H-NMR、13C-NMR等波谱分析鉴定结构,经UHPLC-CAD面积归一化法测得纯度均大于98.0%,结构式如图1所示。
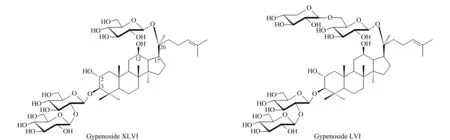
图 1 绞股蓝皂苷XLVI和LVI的结构式Fig. 1 Structures of gypenoside XLVI and LVI

图 2 绞股蓝样品的(a) UHPLC-CAD分析色谱图和(b) UHPLC-Q-TOF/MS分析基峰图Fig. 2 (a) Chromatogram of UHPLC-CAD and (b) base peak ion chromatogram of UHPLC-Q-TOF/MS of Gynostemma pentaphyllum sample Peak identifications 1-8 were shown in Table 2. CAD: charged aerosol detector; Q-TOF/MS: quadrupole time-of-flight tandem mass spectrometry.
药材来源信息见表1, 16份样品均来自福建绞股蓝主产区,经中国医学科学院药用植物研究所郭宝林研究员鉴定为葫芦科绞股蓝属植物绞股蓝。所有药材经烘箱50 ℃干燥至恒重后,粉碎,过40目筛得样品粉末,干燥阴凉处储存备用。
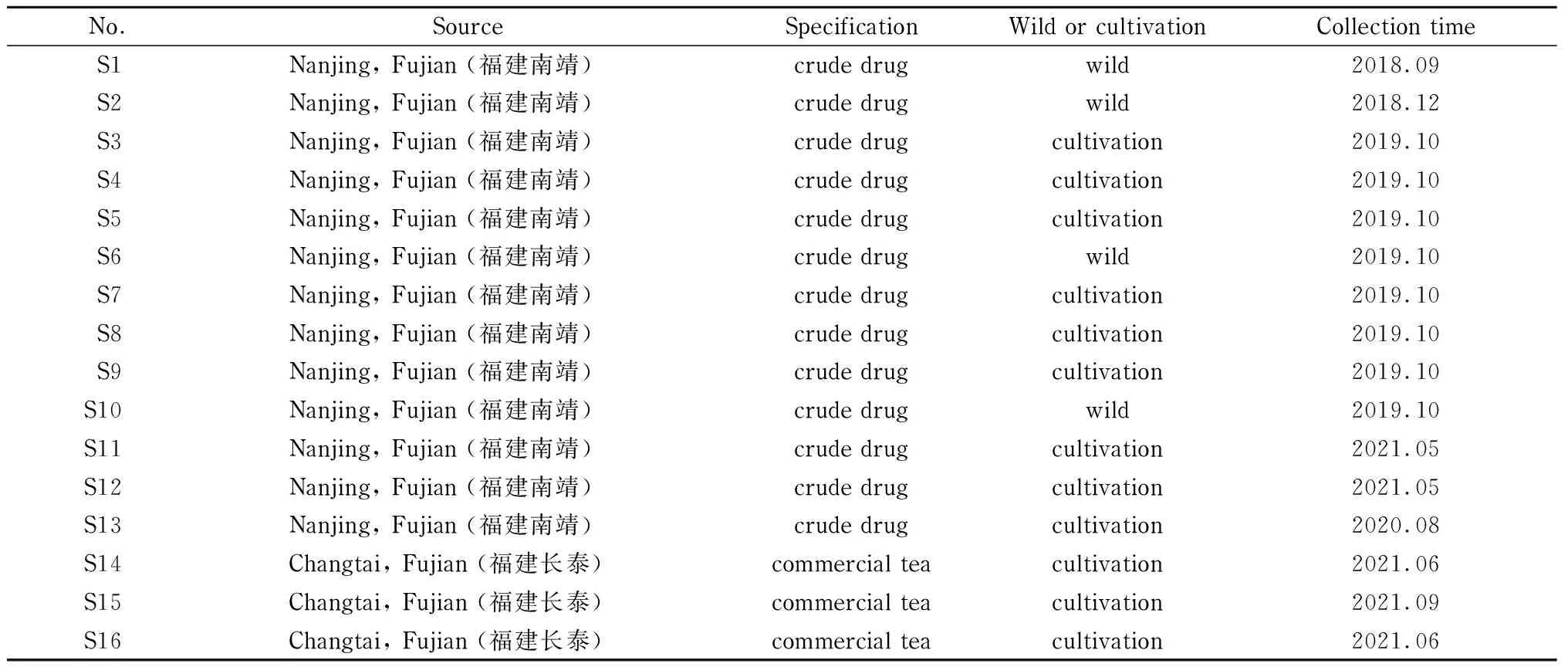
表 1 绞股蓝样品信息Table 1 Information of Gynostemma pentaphyllum samples
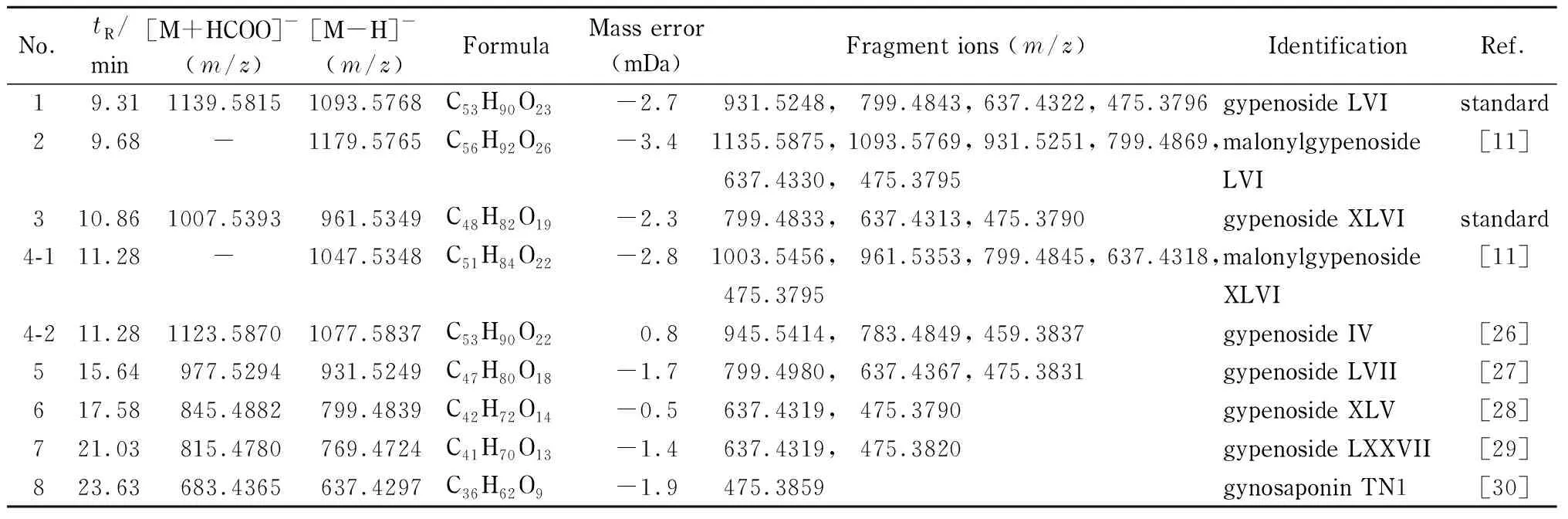
表 2 绞股蓝样品中主要的化学成分Table 2 Main compounds in Gynostemma pentaphyllum samples
1.2 定性分析
1.2.1定性分析样品的配制
称取绞股蓝皂苷XLVI和LVI对照品粉末适量,分别溶解于乙醇-水(70∶30, v/v)中,各吸取一定体积均匀混合,0.22 μm微孔滤膜过滤即得对照品溶液。
精密称取绞股蓝样品粉末0.2 g,加入乙醇-水(70∶30, v/v)30 mL于具塞锥形瓶中,超声提取30 min,取上清液经0.22 μm微孔滤膜过滤即得供试品溶液。
1.2.2定性分析的液相色谱条件
色谱柱:Waters ACQUITY UPLC BEH C18色谱柱(100 mm×2.1 mm, 1.7 μm);流动相A为0.1%(v/v)甲酸水溶液,流动相B为乙腈。梯度洗脱条件:0~2 min, 10%B~25%B; 2~7 min, 25%B~32%B; 7~10 min, 32%B~33%B; 10~12 min, 33%B~35%B; 12~22 min, 35%B~46%B; 22~27 min, 46%B~56%B; 27~28.5 min, 56%B~95%B; 28.5~30 min, 95%B; 30~30.5 min, 95%B~10%B; 30.5~33 min, 10%B。流速为0.5 mL/min;柱温为40 ℃;进样体积2 μL。
1.2.3飞行时间质谱条件
电喷雾电离(ESI)源,负离子模式,离子源温度为110 ℃,脱溶剂气体为氮气,流速为850 L/h,温度为450 ℃,毛细管电压为2.5 kV,锥孔电压为50 V,低能量扫描时能量为6 eV,高能量扫描时能量为35~65 eV,扫描范围为m/z100~1 500。精确质量数用亮氨酸-脑啡肽(leucine-enkephalin)溶液进行校正。
1.3 含量测定
1.3.1对照品溶液的配制
分别精密称取对照品粉末绞股蓝皂苷XLVI和LVI适量,用乙醇-水(50∶50, v/v)溶解,于10 mL容量瓶中定容,制得质量浓度分别为1.272 mg/mL和1.636 mg/mL的对照品储备溶液,4 ℃冰箱保存备用。
1.3.2供试品溶液的配制
精密称取样品粉末0.2 g,置于100 mL具塞磨口锥形瓶中,加入30 mL乙醇-水-氨水(50∶46∶4, v/v/v),密塞,超声提取30 min(功率600 W,频率40 kHz),静置24 h,称重比较前后重量差异,加入提取溶剂补足减失的量,取1 mL上清液经0.22 μm微孔滤膜过滤,待测。
1.3.3含量测定的液相色谱条件
色谱柱:Waters ACQUITY UPLC BEH C18色谱柱(100 mm×2.1 mm, 1.7 μm);流动相A为0.1%(v/v)甲酸水溶液,流动相B为乙腈。梯度洗脱条件:0~10.4 min, 30%B; 10.4~10.5 min, 30%B~95%B; 10.5~14.5 min, 95%B; 14.5~14.6 min, 95%B~30%B; 14.6~20 min, 30%B。流速为0.5 mL/min;柱温为40 ℃;进样体积2 μL; 电雾式检测器采集频率10 Hz,蒸发温度35 ℃,滤波1 s。
2 结果与讨论
2.1 主成分鉴定
为充分了解福建产绞股蓝的化学成分信息,利用UHPLC-CAD分析福建产绞股蓝,得到其CAD指纹图谱,如图2a所示,发现色谱峰1~4为福建产绞股蓝的主要成分。在相同指纹图谱液相色谱条件下,利用UHPLC-Q-TOF/MS对福建产绞股蓝进行化学成分鉴定,基峰图如图2b所示。通过化合物的保留时间、相对分子质量、化学式、质量数误差、高能量裂解碎片等信息,结合对照品及相关文献报道即可鉴定出各个色谱峰对应的可能的化合物,鉴定结果见表2。
以色谱峰3和4为例,具体鉴定过程如下。色谱峰3在负离子低能量模式下产生加合离子峰m/z1 007.5 393 [M+HCOO]-和准分子离子峰m/z961.534 9 [M-H]-,推断其分子式为C48H82O19,同时,其可在负离子高能量模式下产生碎片离子m/z799.483 3、637.431 3和475.379 0,表明结构中含有3分子六碳糖。经与对照品保留时间、相对分子质量等信息对比确定其为绞股蓝皂苷XLVI。在负离子模式下色谱峰4产生了两组碎片离子(见图3),分别为m/z1 047.534 8 [M4-1-H]-,以及m/z1 077.583 7 [M4-2-H]-和1 123.587 0 [M4-2+HCOO]-的准分子离子峰,推测色谱峰4含有两个化合物,即化合物4-1和化合物4-2,根据其准分子离子峰,分别推断化合物4-1的分子式为C51H84O22(m/z1 047.534 8 [M4-1-H]-),化合物4-2的分子式为C53H90O22(m/z1 077.583 7 [M4-2-H]-)。通过提取准分子离子峰,化合物4-1在负离子高能量模式下产生碎片m/z1 003.545 6、961.535 3、799.484 5、637.431 8、475.379 5,表明其在高能量时连续脱去中性特征碎片m/z44(CO2)、m/z42(C2H2O)和3分子六碳糖。除脱去m/z44(CO2)、m/z42(C2H2O)外,其他碎片与对照品绞股蓝皂苷XLVI一致,结合文献[11]报道推断化合物4-1为丙二酰基-绞股蓝皂苷XLVI。化合物4-2在负离子高能量模式下产生碎片m/z945.541 4、783.484 9、459.383 7,表明其在高能量时连续脱去1分子五碳糖和3分子六碳糖,结合文献报道[26]推断化合物4-2为绞股蓝皂苷IV。因此色谱峰4中包含了在此色谱条件下未能实现分离的化合物丙二酰基-绞股蓝皂苷XLVI和绞股蓝皂苷IV。

表 3 绞股蓝皂苷XLVI与LVI的线性方程、线性范围、相关系数、检出限和定量限Table 3 Linear equations, linear ranges, correlation coefficients (r), LODs and LOQs of gypenoside XLVI and LVI
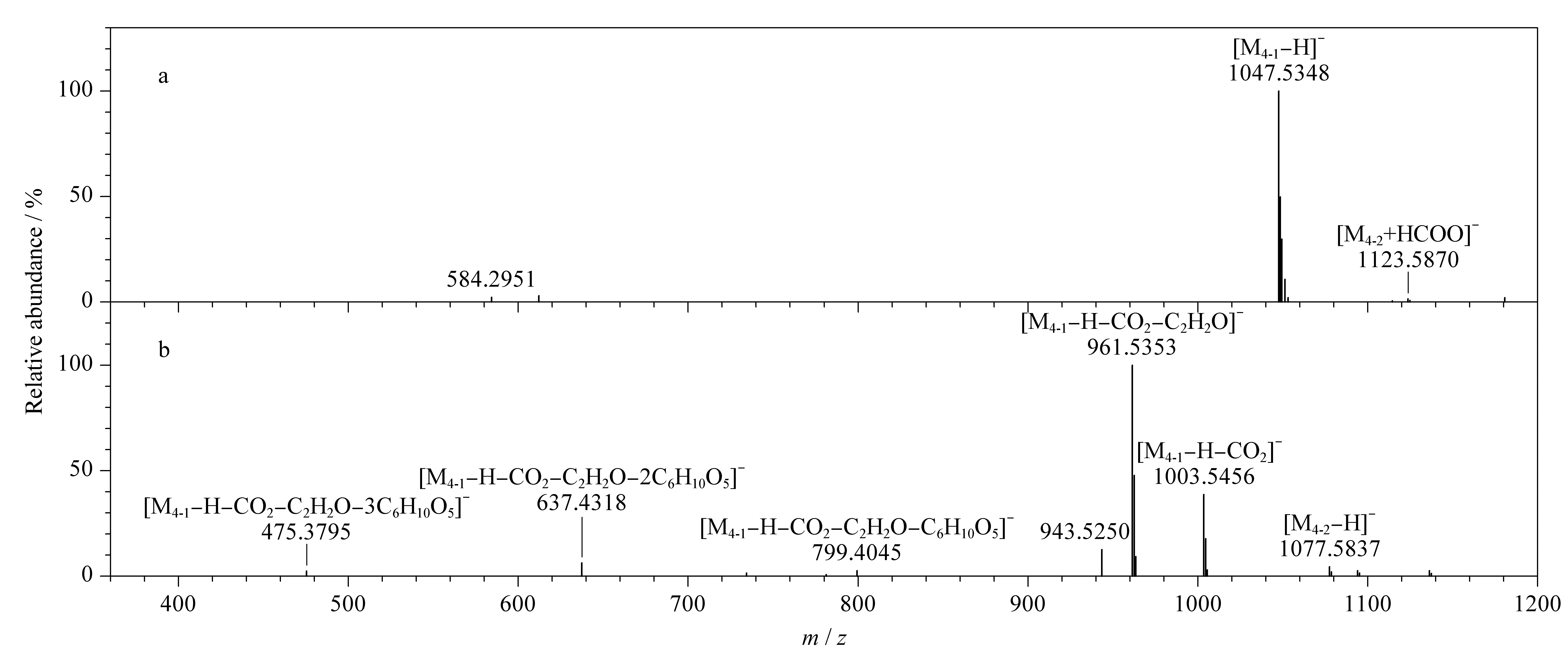
图 3 色谱峰4在负离子(a)低能量和(b)高能量模式下的质谱图Fig. 3 Mass spectra of peak 4 in negative ion (a) low energy and (b) high energy modes
2.2 碱水解条件优化
丙二酰基-绞股蓝皂苷XLVI和LVI在福建产绞股蓝中含量较大,但其不稳定容易脱去丙二酰基,难以得到对照品,故参考人参含量测定文献中对丙二酰基酸性皂苷的处理方法[31],适当进行条件优化后,对绞股蓝样品进行碱水解预处理,再进行含量测定。
分别考察了碱水解中氨水的体积分数(1%、2%和4%)和反应时间(12、24和36 h)对绞股蓝皂苷XLVI、绞股蓝皂苷LVI、丙二酰基-绞股蓝皂苷XLVI和丙二酰基-绞股蓝皂苷LVI含量的影响。当氨水体积分数为1%和2%时,丙二酰基-绞股蓝皂苷XLVI和丙二酰基-绞股蓝皂苷LVI在36 h内均未能完全转化,耗时过长;当氨水体积分数为4%时,24 h内丙二酰基-绞股蓝皂苷XLVI和LVI即可完全转化,且36 h与24 h无明显变化。综合考虑样品溶液的pH值以及反应时长,确定碱水解条件:氨水体积分数为4%,反应24 h。判断碱水解是否完全的过程如图4所示,分别为该条件下碱水解前、后的UHPLC-CAD色谱图,色谱峰2已完全转化,而峰面积相当的色谱峰4没有完全转化,由2.1节已知色谱峰4中包含了2个化合物,其中化合物4-2(绞股蓝皂苷IV)结构中不含有丙二酰基,碱水解对其无影响,结合质谱测定碱水解后该色谱峰m/z1 077.5 837 [M-H]-为化合物4-2,说明含丙二酰基的化合物4-1已全部转化为去丙二酰基的绞股蓝皂苷XLVI,即该条件下碱水解已完全。
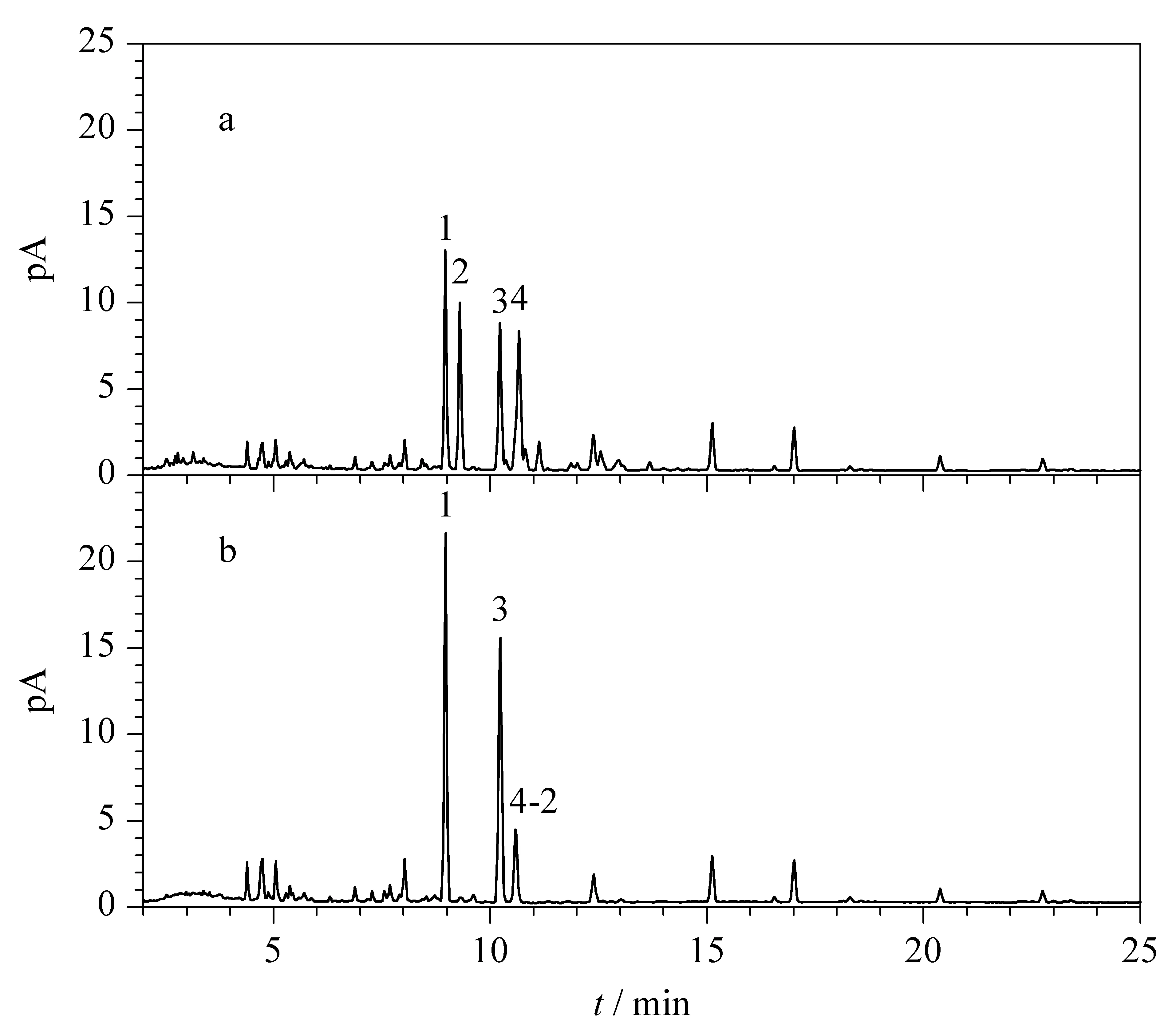
图 4 绞股蓝样品S10在含4%氨水提取溶剂中反应24 h(a)前、(b)后的色谱图Fig. 4 Chromatograms of Gynostemma pentaphyllum sample S10 (a) before and (b) after the reaction in the extraction solvent containing 4% ammonia for 24 h 1. gypenoside LVI; 2. malonylgypenoside LVI; 3. gypenoside XLVI; 4. malonylgypenoside XLVI/gypenoside IV; 4-2. gypenoside IV.
此外,取不同年份、不同来源和不同规格的福建产绞股蓝样品S1、S3、S6和S16在上述条件下进行碱水解,反应后分别取样品适量,在1.2.2节液相色谱条件下进行UHPLC-CAD分析。如图5所示,不同批次福建产绞股蓝样品的一致性较好,该碱水解条件下不同含量的丙二酰基-绞股蓝皂苷XLVI和LVI均能完全转化为相应的绞股蓝皂苷XLVI和LVI,且转化后绞股蓝皂苷XLVI和LVI的总含量远高于其他皂苷成分,进一步验证了优化后的碱水解条件的通用性以及质量评价指标成分选择的合理性。
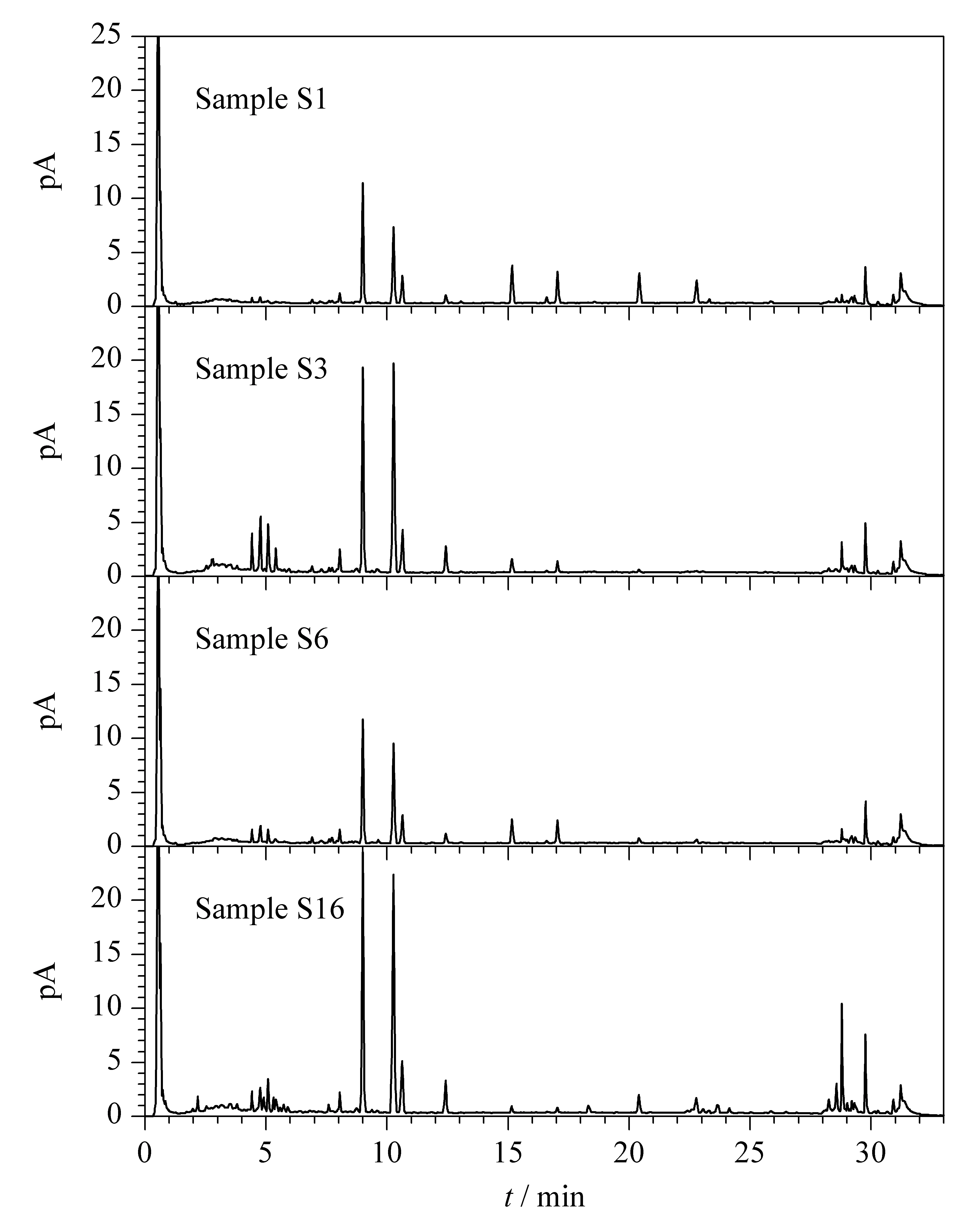
图 5 碱水解后不同批次绞股蓝样品的UHPLC-CAD色谱图Fig. 5 UHPLC-CAD chromatograms of different batches of Gynostemma pentaphyllum sample after the alkali hydrolysis
2.3 提取方法优化
在样品提取的过程中,分别考察了不同提取溶剂、提取料液比以及提取时间对绞股蓝皂苷XLVI和LVI提取率的影响。同时,结合2.2节碱水解条件优化结果,在提取方法优化过程中均在提取溶剂中加入体积分数为4%的氨水。当料液比为1∶150(g∶mL)、超声提取时间为30 min时,在不同提取溶剂(30%、50%、70%甲醇水溶液和30%、50%、70%乙醇水溶液)下,总提取液中绞股蓝皂苷XLVI和LVI的峰面积之和分别为13 378、22 305、24 015、17 670、23 959和21 043,其中,70%甲醇水溶液与50%乙醇水溶液对两种成分的提取率相当,但由于乙醇更加绿色环保,故选用50%乙醇水溶液(含4%氨水)作为提取溶剂。
当提取溶剂为50%乙醇水溶液(含4%氨水),提取时间为30 min时,在不同料液比(1∶50、1∶100、1∶150和1∶200, g∶mL)下,总提取液中绞股蓝皂苷XLVI和LVI的峰面积之和分别是18 152、20 721、23 959和23 974, 1∶150(g∶mL)与1∶200(g∶mL)料液比下,两种成分的提取率相当,综合考虑样品浓度等因素,确定料液比为1∶150(g∶mL)。
此外,当提取溶剂为50%乙醇水溶液(含4%氨水),提取料液比为1∶150(g∶mL),超声提取时间分别为20、30、40和50 min,总提取液中绞股蓝皂苷XLVI和LVI的峰面积之和分别是22 888、23 959、23 917和23 983,当提取时间为30 min时,两种化合物已被充分提取,再随着提取时间的增加,峰面积无明显差异,为缩短分析操作时间,提高分析效率,故选择提取时间为30 min。
最终提取条件确定为:乙醇-水-氨水(50∶46∶4, v/v/v)作为提取溶剂,料液比1∶150(g∶mL),超声提取30 min。
2.4 液相色谱条件的优化
考察了不同的色谱柱以及不同的流动相组成对供试品或对照品峰形和分离效果的影响。由于提取溶液中加入了氨水,供试品溶液具有一定的碱性(pH=8.8),故选择的色谱柱须耐受一定的碱性,选择Waters ACQUITY UPLC BEH C18色谱柱(100 mm×2.1 mm, 1.7 μm; pH耐受范围:1~12)。
关于流动相的组成,考察了不同的流动相系统(水-甲醇和水-乙腈)对化学成分分离效果的影响,结果显示在水-乙腈系统下,各待测成分分离效果较好,响应较高,同时,在实验中发现加入适量的甲酸有助于峰形的改善,因此最终确定流动相A为0.1%(v/v)甲酸水溶液,流动相B为乙腈。如图6所示,该液相色谱条件下供试品中绞股蓝皂苷XLVI和LVI保留适中,分离度良好。

图 6 (a)绞股蓝样品和(b)混合对照品的UHPLC-CAD色谱图Fig. 6 UHPLC-CAD chromatograms of (a) Gynostemma pentaphyllum sample and (b) mixed reference substances
2.5 方法学考察
2.5.1线性关系、检出限和定量限
精密吸取1.3.2节配制的对照品储备液适量,并将其均匀混合,用50%乙醇逐级稀释,制得绞股蓝皂苷XLVI质量浓度分别为318.00、159.00、79.50、39.75、19.88、9.94 μg/mL,绞股蓝皂苷LVI质量浓度分别为409.00、204.50、102.25、51.13、25.56、12.78 μg/mL的系列混合对照品溶液;精密吸取上述系列混合对照品溶液各2 μL,分别按1.3.3节的液相色谱条件进样测定。CAD是基于雾化-气溶胶的HPLC检测器,其响应值与被测物质的量呈指数关系,一般需经对数转化或用二次函数计算[32]。本文以待测组分质量浓度(μg/mL)的对数值(X)为横坐标,峰面积的对数值(Y)为纵坐标建立标准曲线。结果如表3所示,绞股蓝皂苷XLVI在9.94~318.94 μg/mL、绞股蓝皂苷LVI在12.78~409.00 μg/mL范围内,X与Y线性关系良好,相关系数(r)分别为0.999 3和0.999 5。
分别对2种对照品储备液逐级稀释,进样检测,分别以信噪比(S/N)等于3和10为标准,确定2种化合物的检出限(LOD)和定量限(LOQ),其中,绞股蓝皂苷XLVI的检出限为1.58 μg/mL,定量限为6.36 μg/mL;绞股蓝皂苷LVI的检出限为2.05 μg/mL,定量限为8.18 μg/mL。
2.5.2精密度试验
取混合对照品溶液,按1.3.3节的液相色谱条件连续进样6次,记录峰面积。绞股蓝皂苷XLVI和LVI峰面积的RSD值分别为0.88%和0.44%(n=6),表明仪器精密度良好。
2.5.3稳定性试验
取1.3.2节方法制得的同一供试品溶液适量(编号:S10),分别在室温下放置0、2、4、6、8、12、24 h,按1.3.3节的液相色谱条件进样检测,记录峰面积,计算绞股蓝皂苷XLVI和LVI的含量。二者的RSD值分别为0.98%和0.90%(n=6),表明供试品溶液在24 h内稳定。
2.5.4重复性试验
精密称取6份同一批次的绞股蓝样品粉末(编号:S10)0.20 g,按1.3.2节方法制备供试品溶液,按1.3.3节的液相色谱条件连续进样,记录峰面积,计算绞股蓝皂苷XLVI和LVI的含量。二者RSD值分别为1.39%和1.55%(n=6),表明该方法的重复性良好。
2.5.5加标回收率试验
精密称取6份同一批次的绞股蓝粉末(编号:S10)0.10 g,精密加入相当量的对照品溶液,按1.3.2节方法制备供试品溶液,按1.3.3节液相色谱条件,连续进样检测,记录峰面积,计算绞股蓝皂苷XLVI和LVI的含量。绞股蓝皂苷XLVI与LVI的加标回收率分别在100.2%~107.2%与97.9%~104.2%范围内,RSD值分别为2.4%与2.6%,表明该方法准确性良好。
2.6 绞股蓝样品含量测定
对包括种植、野生和商品茶在内的16批福建产绞股蓝样品进行含量测定,分别精密称取样品粉末0.2 g,按1.3.2节方法制备供试品溶液,按1.3.3节液相色谱条件进样检测,分别测定各批次绞股蓝样品中绞股蓝皂苷XLVI和LVI的含量,结果见表4。绞股蓝中绞股蓝皂苷XLVI的含量在0.57~2.57%之间,绞股蓝皂苷LVI的含量在0.66~2.99%之间。其中,野生绞股蓝样品(编号:S1、S2、S6和S10)中指标成分含量略低于种植样品;商品茶样品(编号:S14、S15和S16)的指标成分含量高于其他样品。
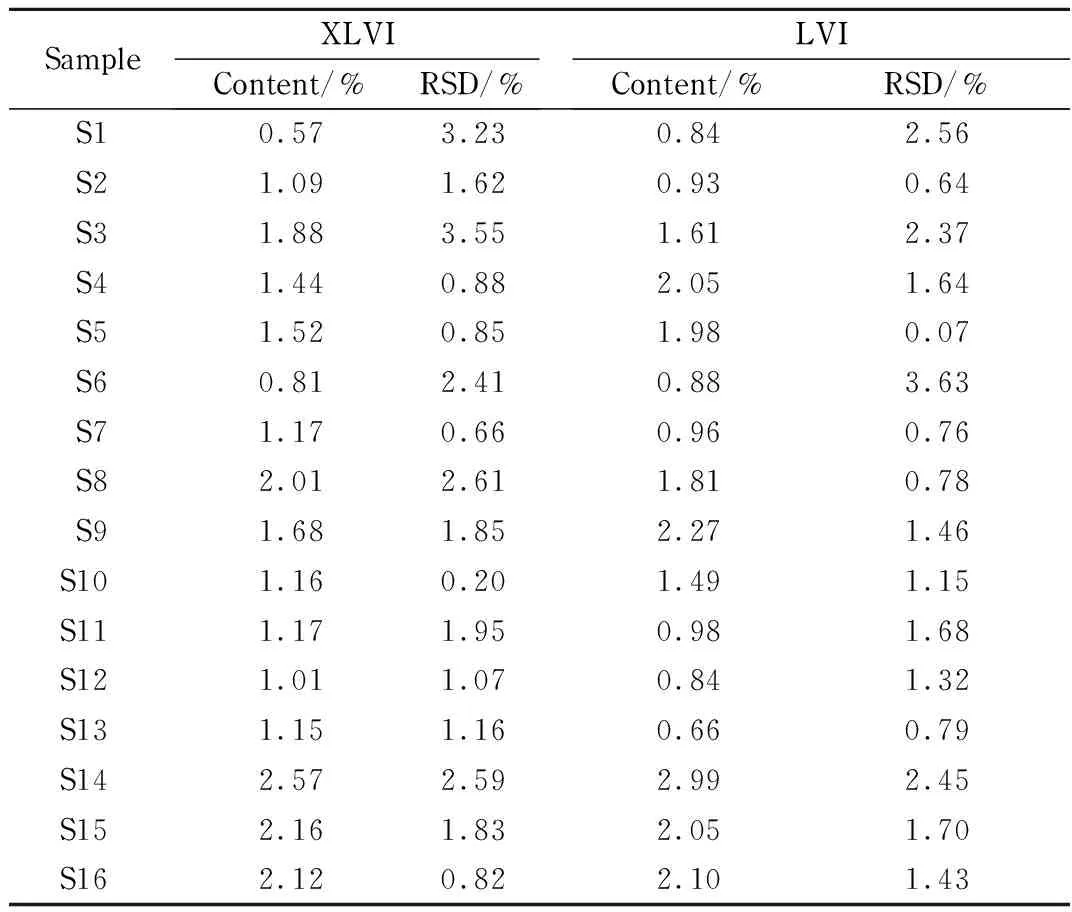
表 4 不同批次绞股蓝样品中绞股蓝皂苷XLVI和LVI的含量(n=3)Table 4 Contents of gypenoside XLVI and LVI in different batches of Gynostemma pentaphyllum samples (n=3)
3 结论
本研究利用UHPLC-Q-TOF/MS结合UHPLC-CAD鉴定了福建产绞股蓝的主要成分,选择2个含量较高的共有成分绞股蓝皂苷XLVI和LVI作为指标成分,利用UHPLC-CAD建立了福建产绞股蓝中绞股蓝皂苷XLVI和LVI含量的测定方法。该方法灵敏度高,重复性好,可用于福建产绞股蓝的质量评价和质量控制,为福建产绞股蓝质量标准的建立和完善提供了方法参考。
致谢 感谢安康正大制药有限公司的李金灿、张军、师东晓协助采集和搜集绞股蓝样品。
猜你喜欢
世界科学技术-中医药现代化(2021年9期)2021-12-31
农业工程学报(2021年3期)2021-04-15
茶叶(2020年1期)2020-05-30
太原理工大学学报(2019年2期)2019-03-13
无机化学学报(2019年2期)2019-02-27
中成药(2017年9期)2017-12-19
恋爱婚姻家庭·养生版(2017年12期)2017-12-07
恋爱婚姻家庭(2017年36期)2017-07-22
中成药(2017年6期)2017-06-13
陕西画报(2016年1期)2016-12-01
