
天津地区寄生烟粉虱的桨角蚜小蜂分子鉴定和遗传多样性分析
2022-09-22 03:30杨泽众王富平胡明鑫徐维红白义川谷希树郭兆将焦克龙刘佰明
环境昆虫学报 2022年4期
张 楠,杨泽众,王富平,胡明鑫,王 芳,徐维红,白义川,谷希树,郭兆将,焦克龙,刘佰明*
(1.天津农学院园艺园林学院,天津 300392;2.天津市农业科学院植物保护研究所,天津 300384;3.中国农业科学院蔬菜花卉研究所,北京 100081)
烟粉虱Bemisiatabaci是重大外来入侵性害虫,广泛为害世界范围内600多种植物。主要通过直接取食、分泌蜜露导致煤污病、传播超过100多种植物病毒等,给作物种植带来巨大的经济损失(王恩东等,2020)。烟粉虱的防治目前仍严重依赖化学防治手段,其优点是能够快速杀死烟粉虱,但长期使用化学杀虫剂,导致烟粉虱抗药性增加,降低防治效果,且带来严重的农药残留问题(Zhangetal.,2020)。应用环境友好型的生防天敌替代化学农药,具有安全、环保等优点,其中寄生蜂作为一类有效的生防天敌,广泛用于防治烟粉虱(王竹红等,2010)。
粉虱寄生蜂主要有恩蚜小蜂属Encarsia和桨角蚜小蜂属Eretmocerus(孟祥锋等,2006),其中恩蚜小蜂属的丽蚜小蜂Encarsiaformosa已经可以商品化生产并大量应用。但是前人研究发现,丽蚜小蜂偏好寄生于温室白粉虱Trialeurodesvaporariorum,桨角蚜小蜂偏好寄生烟粉虱,并取食烟粉虱若虫,对烟粉虱防治有显著成效(戴鹏等,2014;尹园园等,2017)。桨角蚜小蜂是粉虱类害虫的专性寄生蜂,因其触角膨大成桨状而与其它寄生蜂形成显著区别。桨角蚜小蜂均可在粉虱类若虫体内产卵,最喜寄生烟粉虱2龄、3龄若虫(窦文珺等,2020)。通常,单一的寄生蜂产品在不同地区应用过程中,会产生环境不适应现象。挖掘本地寄生蜂资源防治本地害虫是国际通用的生物防治策略。天津地区烟粉虱寄生蜂资源丰富,本研究从天津5个地区初步调查发现天津烟粉虱寄生蜂以桨角蚜小蜂为主,但是粉虱寄生蜂个体较小,从形态上难于精确鉴定,为进一步研究应用造成很大障碍。
分子标记技术可以快速准确的鉴定寄生蜂种类,并进一步明确其系统发育关系(邱宝利等,2005)。利用分子标记手段进行寄生蜂鉴定大体分为核糖体 28S的D2和D3扩展区序列测定并进行不同物种间的同源性比较(薛夏等,2012)、筛选寄生蜂特定引物的SCAR标记技术(张锐锐等,2012)和基于线粒体mtDNACOI基因构建系统发育树鉴定不同地理种群(谢艳兰等,2019)、利用线粒体mtDNACOI基因PCR方法扩增某一基因片段区别亲缘关系较近的寄生蜂种类并鉴定寄生蜂发育情况(张晓曼等,2013)等几类,其中线粒体mtDNACOI基因是应用最普遍的分子标记手段。De Barroetal.(2000)通过对COII、ITS1、ITS2、28S的D2和D3基因片段的研究结合形态学方法描述了澳大利亚寄生烟粉虱及温室白粉虱的3种桨角蚜小蜂种群。Montietal.(2005)通过测序和PCR-RFLP两种方法研究了线粒体mtDNACOI基因的1~900 bp区段DNA变异情况,成功鉴定了形态学上难于区分的Encarsiaformosa、Encarialuteola和来自巴基斯坦及西班牙的两个Encarsiasophia种群。张毅波等(2016)基于线粒体mtDNACOI基因序列构建系统发育树和种间杂交的方法成功鉴定了廊坊和新疆的桨角蚜小蜂种群,其中桨角蚜小蜂廊坊种群与古桥桨角蚜小蜂Eretmocerusfuruhashii聚为一支,桨角蚜小蜂新疆种群与海氏桨角蚜小蜂Eretmocerushayati聚为一支。通过杂交实验得出,新疆种群与海氏桨角蚜小蜂无生殖隔离,可能属于同一物种。
本研究利用线粒体mtDNACOI基因作为分子标记,对天津市5个地区13个种群的粉虱寄生蜂COI基因进行测序,在分子水平上分析桨角蚜小蜂种属间的系统发育关系,对天津桨角蚜小蜂的遗传多样性进行研究及桨角蚜小蜂发生扩散提供理论依据,为进一步鉴定、开发应用天津地区本地烟粉虱寄生蜂提供参考。
1 材料与方法
1.1 供试虫源
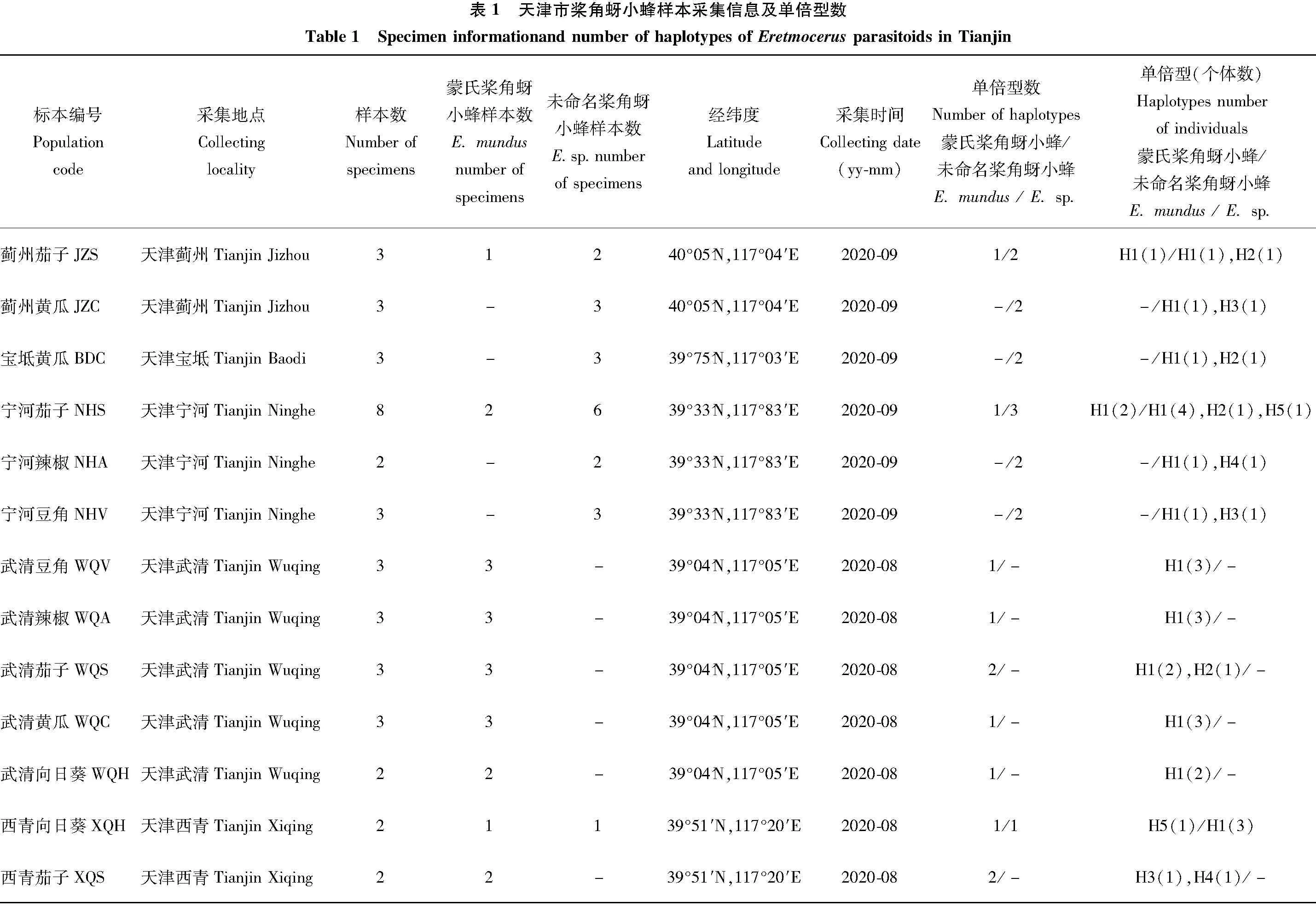
于2020年8-9月在天津市蓟州、宝坻、宁河、武清、西青5个地区采集了13个桨角蚜小蜂种群,用直径5 mm长35 mm的小玻璃管采集植物叶片上桨角蚜小蜂成虫,转入盛有75%酒精的1.5 mL离心管中,标明采集地点、寄主植物、经纬度和采集时间等,保存于4℃冰箱内备用。具体样本信息见表1。
1.2 DNA提取
使用KAPA DNA提取试剂盒进行单头桨角蚜小蜂DNA提取。用昆虫针挑取一头寄生蜂至2 mL离心管中,加入2 mm研磨钢珠,再加入KAPA DNA提取试剂盒体系30 μL(张邓壮等,2020),使用G100高通量组织破碎仪1 800 rpm研磨1 min,8 000 rpm离心30 S,吸28 μL上清至新PCR管中。反应程序为:75℃裂解10 min,95℃ 5 min。
1.3 PCR扩增及序列
PCR扩增引物为:C1-J-2183:5′-CAACATTTA TTTTGATTTTTTGG-3′和L2-N-3014:5′-TCCAATG CACTAATCTGCCATATTA-3′。反应体系为25 μL,其中超纯水7 μL,2×缓冲液 10 μL(包含镁离子),每种引物1 μL,模板DNA 2 μL(Simonetal.,1994)。扩增程序为:95℃预变性7 min;35个循环:95℃ 30 S,55℃ 30 S,72℃ 1 min;最后72℃延伸7 min。扩增产物为COI基因3′末端的部分序列,大小为840 bp左右。取4 μL PCR产物于1.5%琼脂糖凝胶电泳检测。扩增成功的PCR产物送往北京生物工程技术服务有限公司测序。
1.4 序列分析
所得PCR扩增产物序列用MEGAX(Kumaretal.,2018)软件进行校对编辑和检测,将整理后的序列在NCBI网站上进行BLAST搜索和比对。在MEGAX中的Clustal W进行多重序列比对,计算碱基组成、变异位点和转换/颠换偏倚率等。用Kimura 2-Parameter(K2P)模型分别计算单倍型间的遗传距离。利用DnaSP 5.10(Librado and Rozas,2009)计算桨角蚜小蜂mtDNACOI基因序列单倍型数目和出现频率、单倍型多样度Hd、核苷酸多样度Pi、核苷酸平均差异数K并进行错配分布(Mismatch Distribution)分析。
1.5 系统发育分析
利用Blast在线比对,检索测序结果。分别下载康尼氏桨角蚜小蜂EretmoceruscocoisEU017333等、海氏桨角蚜小蜂EretmocerushayatiKF859858、未命名桨角蚜小蜂WTT-2016Eretmocerussp.WTT-2016 KX714952和蒙氏桨角蚜小蜂EretmocerusmundusJN62721等序列。将下载序列与测序结果混合,并利用mafft软件比对。利用Jmodeltest 2软件计算最佳进化模型。最后,根据计算得到的最佳进化模型,利用Mrbayes 3.0构建进化树(Fredriketal.,2003)。
2 结果与分析
2.1 天津地区桨角蚜小蜂资源调查结果
2020年8月至2020年9月在天津市5个区共采集到40头桨角蚜小蜂。经DNA基因提取和线粒体COI基因扩增测序后,成功得到40条线粒体mtDNACOI基因序列,通过NCBI比对发现采集到的样品与蒙氏桨角蚜小蜂(Eretmocerusmundus,20头)和未命名桨角蚜小蜂(Eretmocerussp.WTT-2016,20头)亲缘关系较近。蒙氏桨角蚜小蜂在武清区分布较多,蓟州、宁河及西青区分布较少,宝坻区未发现;未命名桨角蚜小蜂在宁河区分布较多,蓟州和宝坻区分布较少,武清和西青区未发现(表1)。
2.2 桨角蚜小蜂线粒体mtDNA COI基因的碱基组成及序列变异
蒙氏桨角蚜小蜂共20条线粒体mtDNACOI基因序列,比对后截齐两端得到755 bp的对齐序列。所测序列中保守位点720个,变异位点35个,其中简约信息位点4个,单一变异位点31个,总体转换/颠换偏倚率R值为3.3。所测序列中碱基A、T、G、C的含量分别为35.6%、42.7%、12.7%和9%;A+T含量占78.4%,表现较强的A/T偏倚。
未命名桨角蚜小蜂共20条线粒体mtDNACOI基因序列,比对后截齐两端得到739 bp的对齐序列。所测序列中保守位点735个,变异位点4个,其中简约信息位点1个,单一变异位点3个,总体转换/颠换偏倚率R值为6.8。所测序列中碱基A、T、G、C的含量分别为34.8%、43.8%、12%和9.4%;A+T含量占78.6%,表现较强的A/T偏倚。
2.3 桨角蚜小蜂mtDNA COI基因的单倍型系统发育分析
采用Mrbayes 3.0构建进化树,Jmodeltest 2分析得出最佳进化模型(GTR+I+G)。以丽蚜小蜂Encarsiaformosa线粒体mtDNACOI基因序列(登录号:AY264337)作为外群为代表,包括浅黄恩蚜小蜂Encarsiasophia等蚜小蜂科为代表在NCBI中获得登录号。利用Mrbayes 3.0构建10个桨角蚜小蜂单倍型系统发育树(图1)。可以看出,本研究中桨角蚜小蜂种群(MH1-MH5)与已知蒙氏桨角蚜小蜂种群聚为一大分支,利用MEGA X进一步分析发现两种群内的遗传距离分别为0.02和0.01,种群间遗传距离为0.02。本研究中另一桨角蚜小蜂种群(SPH1-SPH5)与Eretmocerussp.WTT-2016聚为一大支,进一步分析发现两种群内的遗传距离分别为0.00和0.00,种群间遗传距离为0.00。
2.4 桨角蚜小蜂单倍型遗传距离
遗传距离的计算结果表明,蒙氏桨角蚜小蜂线粒体mtDNACOI基因5个单倍型间的遗传距离为0.0013~0.0438(表2),种内平均遗传距离为0.02。未命名桨角蚜小蜂线粒体mtDNACOI基因5个单倍型间的遗传距离为0.0014~0.0041(表2),种内平均遗传距离为0.00。

表2 蒙氏桨角蚜小蜂(下三角)和未命名桨角蚜小蜂(上三角)mtDNA COI基因5个不同单倍型间的遗传距离Table 2 Genetic distance among 5 different haplotypes of mtDNA COI gene of Eretmocerus mundus(below the diagonal) and Eretmocerus sp.WTT-2016 (above the diagonal)
2.5 桨角蚜小蜂mtDNA COI基因的单倍型和遗传多样性分析
共享单倍型为种群内大多数个体共有的单倍型,独享单倍型为种群内单独个体有的单倍型。蒙氏桨角蚜小蜂种群中共获得线粒体mtDNACOI基因的5个单倍型MH1-MH5,其中MH1为共享单倍型,MH2-MH5单倍型为相应种群的独享单倍型;共享单倍型MH1分布最广占蒙氏桨角蚜小蜂检测个体的70%。未命名桨角蚜小蜂种群中共获得mtDNACOI基因的5个单倍型SPH1-SPH5,其中SPH1-SPH3为共享单倍型,SPH4、SPH5单倍型为相应种群的独享单倍型;共享单倍型中SPH1分布最广占未命名桨角蚜小蜂检测个体的60%。
利用DnaSP 5.10计算得出蒙氏桨角蚜小蜂群体单倍型遗传多样度Hd为0.368 ± 0.0185,核苷酸多样度Pi为0.00557 ± 0.0000129,核苷酸平均差异数K为4.205。未命名桨角蚜小蜂群体单倍型遗传多样度Hd为0.616 ± 0.01132,核苷酸多样度Pi为0.00106 ± 0.0000001,核苷酸平均差异数K为0.784。蒙氏桨角蚜小蜂单倍型遗传多样度Hd比未命名桨角蚜小蜂低,Pi和K比未命名桨角蚜小蜂高。
2.6 桨角蚜小蜂错配分析
错配分析(Mismatch Analysis)可以看出桨角蚜小蜂群体在天津发展的历史动态,错配分析图为多峰曲线,表明此种群存在时间较长且近年来未出现扩张现象;错配分析图为单峰曲线,则表明此种群形成时间较短且出现扩张现象(Pengetal.,2017)。从错配分析图可以看出,多峰为蒙氏桨角蚜小蜂,单峰为未命名桨角蚜小蜂(图2)。

图2 基于mtDNA COI基因的蒙氏桨角蚜小蜂和未命名桨角蚜小蜂错配分布分析Fig.2 Mismatch distribution analysis of populations of Eretmocerus mundus and Eretmocerus sp.WTT-2016 based on mtDNA COI gene
3 结论与讨论
桨角蚜小蜂个体较小,形态相似,不易捕捉,一般的形态学方法难以区分近缘物种。分子生物学技术可以快速鉴定物种,Hebertetal.(2003)提出利用mtDNACOI基因分子标记鉴定物种必须满足两个条件:即两个物种的种间基因差异应远大于种内差异;同时种间差异小于0.02时,可以推断两者为同种。本研究通过构建桨角蚜小蜂单倍型系统发育树,所有种形成明显分支,桨角蚜小蜂种群(MH1-MH5)与蒙氏桨角蚜小蜂聚为一支,蒙氏桨角蚜小蜂种间遗传距离为0.02,可初步推断为同种。本研究中另一桨角蚜小蜂种群(SPH1-SPH5)与Eretmocerussp.WTT-2016聚为一支,未命名桨角蚜小蜂种间遗传距离为0.00,可推断为同种。
本研究获得蒙氏桨角蚜小蜂Eretmocerusmundus和未命名桨角蚜小蜂Eretmocerussp.WTT-2016共40条线粒体mtDNACOI基因序列,碱基组成表现较强的A/T偏倚性,与典型的昆虫线粒体碱基组成特点一致(赵乐等,2018)。蒙氏桨角蚜小蜂总的转换/颠换(R)值为3.3,未命名桨角蚜小蜂R值为6.8,都属于核苷酸转换大于颠换,表明蒙氏桨角蚜小蜂不同地理种群亲缘关系较近,未命名桨角蚜小蜂不同地理种群亲缘关系较近(Simonetal.,1994)。此次序列分析定义了5种蒙氏桨角蚜小蜂单倍型和5种未命名桨角蚜小蜂单倍型,两种小蜂单倍型中都是H1发生频率最高、分布最广,可认为是较为原始、能够适应环境变化,并在种群中稳定存在的优势单倍型。两种桨角蚜小蜂都拥有共享单倍型和独享单倍型,表明各个地理种群之间既有基因交流,又存在遗传分化的现象(孙嵬等,2013;刘晓娜等,2016)。
本研究结果显示蒙氏桨角蚜小蜂错配分析图为多峰曲线(图2),表明此种群存在时间较长且近年来未出现扩张现象;未命名桨角蚜小蜂为单峰曲线(图2),表明此种群形成时间较短且出现扩张现象(Pengetal.,2017)。蒙氏桨角蚜小蜂和未命名桨角蚜小蜂的总群体单倍型指数Hd分别为0.368和0.616,表明未命名桨角蚜小蜂比蒙氏桨角蚜小蜂线粒体mtDNACOI基因具有较高的多态性(谢艳蓝等,2019),综上所述,可以推断出蒙氏桨角蚜小蜂种群在天津地区稳定存在,未命名桨角蚜小蜂种群存在时间较短且具有较强的适应能力及遗传变异能力。
本研究在天津各地区采样地点和采集数量较少,初步调查发现天津市5个地区存在桨角蚜小蜂,西青区和宁河区发现少量恩蚜小蜂,后续应增加采集数量及采样点并检测每年天津寄生蜂的种类变化。仅以线粒体mtDNACOI基因作为分子标记并检测,在种群遗传多样性和遗传变异等分析上有偏差。天津桨角蚜小蜂的遗传变异及种群多样性应分析深层原因,加大采样点并结合更多分子标记进行系统深入研究。
猜你喜欢
医学研究生学报(2022年5期)2022-12-07
临床肺科杂志(2022年3期)2022-11-26
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国森林病虫(2022年1期)2022-02-19
学苑创造·A版(2022年2期)2022-01-29
科学之友(2021年12期)2021-12-23
中国卒中杂志(2021年7期)2021-11-29
蜜蜂杂志(2019年12期)2019-06-16
小学生作文选刊·中高年级版(2017年4期)2017-04-24
山东农业科学(2015年3期)2015-05-06
