
纤维肉瘤型隆突性皮肤纤维肉瘤伴肺转移1例报告
2022-09-19 12:27:00何璋海李宝璇李海刚
实用癌症杂志 2022年9期
何璋海 李宝璇 李海刚 王 林
隆突性皮肤纤维肉瘤(dermatofibrosarcoma protuberans,DFSP)是一种发生在真皮层及皮下组织、发病率较低的低度恶性潜能的肿瘤,具有局部侵袭性且容易复发的特点[1],但极少发生转移[2]。DFSP按组织学可分为10多个亚型,其中以纤维肉瘤型(fibrosarcomatous dermatofibrosarcoma protuberans,FS-DFSP)最容易复发,侵袭性最强。现报告1例伴肺转移的纤维肉瘤型隆突性皮肤纤维肉瘤。
1 病例报告
患者男性,60岁,因发现左肩锁关节肿物2年余入住中山大学孙逸仙纪念医院,2年来肿块无明显增大,无明显不适,周围皮肤红绀。
2018年4月,首次入院时查体左肩关节触及一肿块,肿块表面皮肤红肿,活动度欠佳。MRI检查:左肩锁关节后上部皮下脂肪层内见一梭形肿块,大小约64 mm×39 mm×42 mm,内见条索状T1W1高T2W1压脂低信号分割,考虑偏良性病变,神经源性或血管瘤肿瘤可能性大。行肿块手术单纯切除,术后病理检查:左肩部原发肿物,巨检:皮下见一结节状肿物,大小约7 cm×4.7 cm×2.6 cm,切面灰红灰白色,实性,部分质软,部分质中,边界欠清。镜检:浸润皮下脂肪组织成蜂窝状,梭形肿瘤细胞稀疏或密集生长,稀疏区呈车辅状或鱼骨状、漩涡状排列,密集区呈长条束状排列或血管外皮瘤样排列(图1A),瘤细胞密集区细胞核深染,核分裂像易见(约23个/10HPF)。免疫组化:P53(+),S-100部分(+)(图1B)、SOX10部分(+),CD34弥漫(+)(图1C),EMA、Actin、Desmin、CD31、HMB45、MelanA均为(-),Ki67约25%(+)。诊断符合恶性间叶源性肿瘤,需要鉴别恶性外周神经鞘瘤、梭形细胞黑色素瘤与隆突性皮肤纤维肉瘤,结合临床及肿物生长方式,倾向于纤维肉瘤型隆突性皮肤纤维肉瘤(ICD-0编码 8832/3)。术后未行放疗和化疗治疗。

图1 A:原发灶HE,瘤细胞呈“鱼骨状”排列;B:原发灶瘤细胞S-100部分(+);C:原发灶CD34弥漫(+)
2018年9月,因肿瘤于原手术部位复发入院。查体:左肩见一隆起型肿物,活动度欠佳,肿块周围皮肤红绀。MRI检查:左肩背部皮肤增厚,皮下脂肪层内见一梭形肿块,大小约22.4 mm×16.1 mm×25.4 mm,T1W1呈等低信号,T2W1呈稍高信号,提示局部肿瘤复发。行肿瘤扩大切除+皮瓣植入术,切除肿块送病理检查。巨检:距皮肤最近切缘约2.5 cm处见一隆起型肿物,大小约2.5 cm×2.5 cm×2 cm,切面灰白灰褐色,鱼肉样,边界欠清,表面皮肤未见溃疡。镜检:肿瘤细胞形态同原发肿物,梭形细胞呈席纹状排列。免疫组化:CD34部分(+),S-100个别(+),Ki67热点区域约35%(+),SOX10、Actin、Desmin、HMB45、MelanA均(-)。诊断符合纤维肉瘤型隆突性皮肤纤维肉瘤复发。术后行左肩局部放疗,放疗剂量60 Gy,30次。
2020年8月,随诊体检,外院肺部CT显示:左肺上叶尖后段见大小约57 mm×52 mm的肿块,见分叶征、毛刺征,内部密度不均匀。左肺见多个结节影,最大径8 mm,右肺见多结节影,最大径3 mm,左肺门和纵膈见多发淋巴结增大,提示肺转移瘤伴左肺门、纵膈淋巴结转移(图2)。行穿刺活检,活检结果:巨检见灰白色穿刺样组织两条,长1.8~2 cm,直径均0.1 cm,镜检:梭形细胞肿瘤,于肺泡间隔中浸润性生长,细胞中度异型,核分裂像多见,可见病理性核分裂像(图3A)。免疫组化:S100(-)(图3B),H3K27me3部分弱(+)、CD34(+)(图3C),SOX10、EMA、STAT6均为(-),P53约70%弱(+),Ki67约15%(+),TTF-1肺泡上皮(+)。诊断符合恶性间叶源性肿瘤,较符合恶性外周神经鞘瘤。
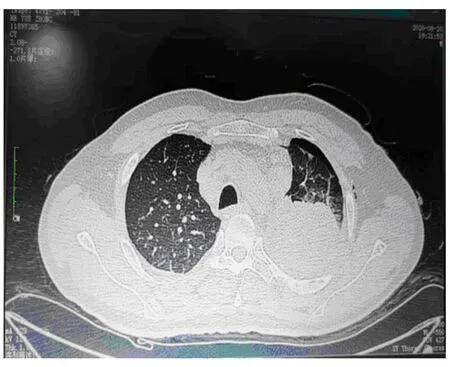
图2 肺转移灶治疗前CT

图3 A:肺转移灶HE,瘤细胞呈“席纹状”排列。B:肺转移灶瘤细胞S-100(-),EnVision法。C:肺转移灶CD34弥漫(+),EnVision法
后行放射粒子消融术治疗,未行化疗,术后CT效果如图4。

图4 肺转移灶治疗后CT
2020年9月行NGS检测。结果如下:COL1A1 exon32-PDGFB exon2 融合(图5),CDKN2A、CDKN2B 基因缺失,RAD51C/ANKFN1重排,RAD51C/CA10重排。

图5 二代基因测序(NGS)显示COL1A1/PDGFB基因融合
2 讨论
DFSP占成人软组织肉瘤的5%,约占全部恶性肿瘤的0.1%[3],而FS-DFSP占DFSP的5%~16%,病理组织学表现为部分区域肿瘤细胞异型性明显,核分裂像增多,见较多病理性核分裂像(>4个/10 HPF);部分瘤细胞排列甚至失去典型席纹状结构,而呈鱼骨状或条束状排列,类似纤维肉瘤。鉴别诊断上,需要与以下肿瘤鉴别:(1)恶性神经鞘瘤:多见于深部组织,与神经关系密切;镜下肿瘤常显示交替性分布的细胞稀疏区和丰富区,稀疏区内血管周围可见瘤细胞聚集,细胞核呈逗点状或波浪状;免疫组化:瘤细胞可表达S-100和SOX10,EMA(+),但相关研究表明H3K27me3的表达缺失,则对恶性神经鞘瘤的诊断具有更高的敏感性和特异性,整体检出率可达65.11%[4],根据其发病原因可分成NF-1相关型、散发型及放疗(radiation therapy,RT)相关型。本病例在二次复发后进行了放疗,但放疗距离发现肺转移病灶尚不足两年,放疗是否导致两种恶性梭形细胞肿瘤的转变或分化还有待考证;(2)恶性纤维组织细胞瘤:目前归入未分化肉瘤,多见于深部软组织,细胞明显异型,常见瘤巨细胞,核分裂像多见,瘤细胞CD34(+),局部可见含铁血黄素成分;(3)浅表性纤维肉瘤:细胞排列呈鲱鱼骨样结构,异型性明显,CD34(-)。
鉴于纤维/肌纤维/纤维组织肿瘤在形态学和免疫表型上均有较多重叠,在难以明确区分的情况下,二代基因测序(NGS)就能在忽略传统病理诊断的差异,提供明确的靶向用药方案。研究发现,DFSP中存在特异性COL1A1/PDGFB 基因融合[5],且检出率达87%,即使在黏液型、纤维肉瘤型、色素型也阳性,对照组阴性的结果也证实了COL1A1/PDGFB 融合基因的表达与病理类型无关[6]。DFSP发病的分子机制是DFSP存在17号染色体和22号染色体易位及额外环状染色体,导致17号染色体上的Ⅰ型A1胶原基因(collagen type Ⅰ a gene,COL1A1)同22号染色体上血小板衍生的生长因子β链基因(platelet-derived growth factor B-chain gene,PDGFB)相融合,发生分子结构重排,产生COL1A1/PDGFB融合基因,该融合基因是DFSP特异性、唯一融合的基因,在其他肿瘤中未检测到该基因。综合本例肿瘤生长特性、组织学、免疫组化以及NGS结果,本例似乎更符合FS-DFSP 的诊断。
COL1A1/PDGFB基因融合是伊马替尼对FS-DFSP靶向治疗的依据[7]。伊马替尼属于酪氨酸激酶抑制剂,治疗主要有这两方面作用:①针对效应T cell调节肿瘤细胞消亡的免疫调节治疗,通过上调抗原提呈作用,以及促进肿瘤微环境中炎症因子和免疫应答相关细胞因子和趋化因子的产生,使肿瘤浸润在细胞毒性T cell中[8];②抑制肿瘤基因激活信号通路。美国食品及药品管理局(FDA)及欧洲药品管理局(EMA)早年已经批准甲磺酸伊马替尼可适用于复发性或转移性的DFSP治疗[9],而对于无法手术切除或切缘阳性的病例,可用伊马替尼治疗后再评估能否手术切除或局部放疗[10],本病例后期已出现肺转移以及纵膈转移,手术难以切除,结合NGS结果,服用伊马替尼。随访至今,患者接受放射粒子治疗2次,规律服用甲磺酸伊马替尼11个月,治疗剂量400 mg/d,随访CT显示:左上肺尖后段不规则软组织密度影,大小2 mm×43 mm,边界不清,纵膈内结构清楚,纵膈、双侧肺门未见明确淋巴结肿大影。随访至今,未见肿瘤复发和转移。
DFSP病理亚型较多且形态相当复杂,CD34瘤细胞弥漫阳性,结合NGS检测COL1A1/PDGFB基因融合,可作为其诊断的重要手段,并为伊马替尼靶向治疗提供依据。
猜你喜欢
中华实用诊断与治疗杂志(2022年2期)2022-09-02 01:47:28
世界最新医学信息文摘(2021年12期)2021-06-09 08:35:02
幸福家庭(2021年1期)2021-03-08 12:31:51
家庭医学(下半月)(2019年11期)2020-01-16 08:39:08
家庭医学(下半月)(2019年10期)2019-11-16 08:59:52
中国临床医学影像杂志(2019年4期)2019-06-18 10:55:06
中国生殖健康(2019年11期)2019-01-07 01:27:38
中国卫生标准管理(2015年1期)2016-01-14 03:41:22
郑州大学学报(医学版)(2015年1期)2015-02-27 14:50:36
肝胆胰外科杂志(2015年4期)2015-02-27 11:12:29
