
酪蛋白激酶-2相互作用蛋白-1介导的自噬通路与骨质疏松症的关系探讨
2022-09-17 09:54:00刘康周航惠明大黄海何才剑房谋昊陈天鹏史晓林
中医正骨 2022年6期
刘康,周航,惠明大,黄海,何才剑,房谋昊,陈天鹏,史晓林
(1.浙江中医药大学附属第二医院,浙江 杭州 310005;2.浙江中医药大学第二临床医学院,浙江 杭州 310053;3.诸暨市中心医院,浙江 诸暨 311899)
骨质疏松症被认为是一种典型的增龄性疾病,其原因是骨重建受损,并伴有成骨细胞和破骨细胞数量和活性的失衡。酪蛋白激酶-2相互作用蛋白-1(casein kinase 2 interacting protein-1,CKIP-1)不仅是细胞存活、凋亡、细胞骨架形成和细胞分化的调节因子,还是骨形成的负性调节因子,导致破骨细胞过度活化和骨丢失。CKIP-1的靶向药物可以促进骨形成,预防骨质疏松症。CKIP-1主要通过介导骨形态发生蛋白(bone morphogenetic protein,BMP)及Wnt两大信号通路来发挥对骨形成的抑制作用。自噬作为一种细胞生存途径,在维持骨稳态方面起着至关重要的作用,而这一途径的变化在一定程度上与骨质疏松的发生有关。与年龄相关的雌激素缺乏被广泛认为是骨质疏松症发生的主要原因,而与衰老相关的骨组织中氧化应激的增加也被认为是骨质疏松症发生的主要致病因素之一。研究发现,CKIP-1能够介导自噬相关通路来影响骨质疏松症的发生发展。靶向调节CKIP-1介导的自噬通路是一种潜在的治疗骨质疏松症的策略。但目前关于CKIP-1介导的自噬通路的相关研究主要集中在肿瘤领域,在骨代谢相关领域的研究较少。本文就CKIP-1介导的自噬通路与骨质疏松症的关系进行了探讨,现报告如下。
1 自噬的概念及发生机制
自噬主要负责不必要的细胞器官和过量营养的循环,以及代谢废物和细胞内病原体的消除。哺乳动物细胞中的自噬可分为3种主要方式:大自噬、微自噬和伴侣蛋白介导的自噬。目前学界对于大自噬(通常所说的自噬)的研究最为深入。在大自噬中,细胞内物质的捕获和传递以自噬小体的形成为标志。在与溶酶体结合后,自噬小体和其所包裹的物质被消化,消化所得产物可被机体进一步重新利用。自噬在生理过程和许多代谢失调相关疾病的发生发展中起到了关键作用。自噬被认为是一种细胞保护机制,能降解异常细胞、细胞器等,但当自噬过度会引起疾病的发生。
包括胰岛素、葡萄糖以及许多生长因子和细胞因子在内的多种分子都可以启动磷脂酰肌醇3-激酶(phosphatidylinositol-3 kinase,PI3K)/蛋白激酶 B/mTOR自噬信号传导。在这些分子的作用下,酪氨酸激酶受体、类Ras蛋白或G蛋白偶联受体等蛋白被激活,随后激活自噬信号级联的触发者PI3K。活化的PI3K可将磷脂酰肌醇-4,5-二磷酸(phosphatidylinositol 4,5-bisphosphate,PIP2)转化为磷脂酰肌醇 -3,4,5-三磷酸(phosphatidylinositol-3,4,5-triphosphate,PIP3),进而激活下游效应因子保证信号的顺利传递。蛋白激酶 B又称 Akt,是 PI3K/Akt/mTOR信号通路中的重要信使。在典型的PI3K/Akt途径中,磷酸肌醇依赖性激酶-1(phosphoinositide-dependent kinase-1,PDK1)和 Akt通过 Pleckstrin同源结构域被募集到细胞膜内表面,其中PDK1在Thr308处启动Akt磷酸化。Akt激活的另一个途径是由mTOR复合物(mammalian target of rapamycin complex,mTORC2)2介导的,它与 Akt的调节疏水结构域相互作用,使其在 Ser473处磷酸化。Akt的一个关键下游分支是mTORC1。磷酸化的Akt可以磷酸化位于Ser2448的mTOR,从而激活mTORC1,也可以磷酸化结节性硬化症复合体2(tuberous sclerosis complex 2,TSC2)间接激活 mTORC1。Akt使 TSC2失活可以抑制TSC1/TSC2复合体的功能,使下游的脑内 Ras同系物(Ras homolog enriched inbrain,RHEB)-GTP转化为 RHEB-GDP,而 RHEB-GTD可导致mTORC1的激活。mTORC1激活后影响其效应物,其中UNC-51样激酶1(unc-51-like kinase 1,ULK1)与自噬的启动相关,mTORC1可以直接磷酸化或抑制ULK1而达到对自噬的抑制。雷帕霉素可导致ULK1去磷酸化,从而使由ULK1、自噬相关基因(autophagy-related genes,ATG)13、ATG101和200 kDa的家族相互作用蛋白(family interacting protein of 200 kDa,FIP200)组成的 ULK1复合体从mTORC1复合体解离。ULK1是已知的自噬相关蛋白中唯一的丝氨酸/苏氨酸蛋白激酶,作为自噬小泡的重要组成成分,与ATG13、ATG101和FIP200形成ULK1复合物而诱导自噬的发生。见图1。

图1 自噬发生机制示意图
2 自噬与骨质疏松症的关系
研究发现,自噬积极参与对骨代谢的调控。自噬对于保持正常成骨细胞的功能是必不可少的,抑制成骨细胞的自噬将导致骨质疏松的发生。研究证实,抑制破骨细胞的自噬在一定程度上能够抑制骨量减少。米健国等认为,去卵巢能够建立绝经后骨质疏松症大鼠模型的原因之一是大鼠血清中丙二醛、活性氧含量增加,过量的活性氧可以抑制PI3K/Akt/mTOR信号通路,激活自噬,而提高自噬水平容易诱发成骨细胞凋亡,进而加速骨量丢失,加快绝经后骨质疏松症的进程。招文华等等研究发现,在激素环境下,前成骨细胞MC3T3-E1成骨分化显著被抑制,mTORC1被下调,自噬激活且Wnt/βcatenin信号通路被抑制,而应用mTORC1激活剂则能逆转该过程,表明 mTORC1能抑制自噬并调控Wnt/β-catenin信号通路介导激素环境下的前成骨细胞MC3T3-E1成骨分化。李希宁等研究发现,通过抑制mTOR磷酸化激活自噬,促进骨形成的同时抑制骨吸收,从而缓解氟骨症大鼠早期骨质疏松。
3 CKIP-1介导的自噬通路与骨质疏松症的关系
CKIP-1在细胞形态、细胞生长发育等方面具有多种生物学功能。而这些功能主要依赖于细胞的位置、类型和调节信号。CKIP-1参与骨形成、肿瘤发生和免疫调节等生物学过程,解除CKIP-1的调控会导致骨质疏松症、肿瘤和动脉粥样硬化的发生。CKIP-1作为骨形成的负调控因子参与骨形成、成骨细胞分化和凋亡等生物学过程,与骨质疏松症的发生密切相关。Zhang等评估了骨组织靶向递送系统“(AspSerSer)-6脂质体”传递的CKIP-1 siRNA的有效性,并检测了骨形态计量学参数、骨量和骨结构;结果发现,该系统传递的CKIP-1 siRNA可以在不影响骨吸收的前提下提高骨形成的能力,表明CKIP-1在逆转绝经后骨质疏松骨丢失方面发挥了重要作用。Liu等研究发现,CKIP-1在骨折患者和衰老啮齿类动物的骨标本中的表达随年龄的增加而增加,这与相关信号的转导及骨形成随年龄的减少有关;还发现CKIP-1 siRNA可以靶向进入成骨细胞,抑制CKIP-1基因表达,从而促进衰老啮齿类动物的骨形成。CKIP-1通过增加Smad泛素化调节因子 -1(Smad ubiquitination regulatory factor-1,SMurf1)与其底物之间的亲和力,增强SMurf1的泛素连接酶活性,促进了Smad1/5的降解,负向调节BMP信号通路而影响骨形成。此外,CKIP-1也能通过Wnt信号受体低密度脂蛋白受体相关蛋白5来影响成骨分化。
CKIP-1在体内和体外均导致细胞生长受阻,是一种新的具有抑制功能的Akt相互作用蛋白。CKIP-1的亮氨酸拉链(leucine zipper,LZ)基序在 Akt激酶活性抑制中起重要作用。CKIP-1可直接与Akt的PH结构域结合,并降低Akt激酶的活性;通过其氨基末端与每个Akt异构体(Akt1、Akt2和Akt3)形成复合物;通过其羟基末端的LZ基序二聚化发现与Akt失活有关,因为LZ基序的缺失消除了Akt抑制功能。尽管LZ基序的缺失使其仍然可以与Akt结合,但通过隔离内源CKIP-1与Akt结合的能力来诱导Akt磷酸化和活化。稳定的CKIP-1表达可导致Akt失活和体外细胞生长抑制。在成骨细胞中,CKIP-1能抑制Akt的磷酸化,上调p-Akt/Akt的比值,而磷酸化的Akt是自噬信号传导过程中的重要一环,磷酸化使Akt激活从而能够转入其他细胞隔间与其下游的相关因子发生作用,最终导致自噬的激活(图2)。自噬激活后抑制成骨细胞分化及生长,导致成骨细胞矿化减少,最终导致骨质疏松的发生。
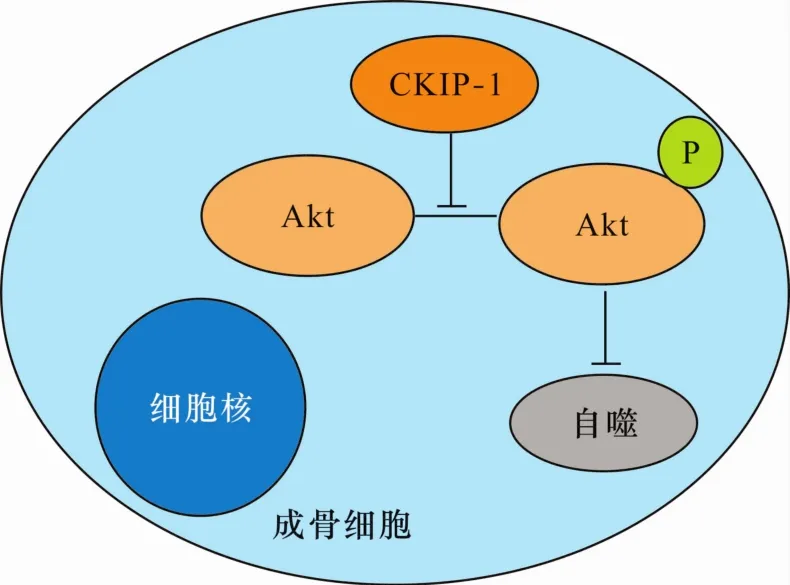
图2 成骨细胞中CKIP-1介导的自噬通路分子机制示意图
Zhang等研究发现,CKIP-1通过与 TRAF6相互作用并抑制Akt激活,CKIP-1缺乏会导致Akt的长时间激活,并且CKIP-1缺陷小鼠自发地出现巨噬细胞所介导的脾肿大和骨髓增殖。Yuan等研究发现,自噬在成骨细胞分化过程中被激活,是因为CKIP-1通过Akt/mTOR信号通路参与骨细胞自噬,CKIP-1与Akt结合而导致该通路受到抑制,从而激活了自噬通路;在使用强骨饮干预后重新激活了被抑制的Akt/mTOR通路,而CKIP-1充当强骨饮和Akt/mTOR信号之间的中介。在体外大鼠成骨、破骨细胞中,CKIP-1可提高成骨、破骨细胞中自噬水平,这也充分证明了CKIP-1介导的自噬通路在骨质疏松症的发生中起着重要作用。
4 小 结
自噬活动与骨代谢之间具有明显的关系,自噬的激活会导致骨代谢的紊乱,而骨代谢的紊乱会直接导致骨质疏松的发生。目前,已有研究采用调节自噬的方式来治疗因衰老、雌激素缺乏或使用糖皮质激素而引起的骨质疏松症,并取得了良好的疗效。CKIP-1主要通过与Akt结合,抑制Akt磷酸化使其失活,并使PI3K/Akt/mTOR信号传导受阻来激活自噬,进而抑制骨分化及生长,最终导致骨质疏松的发生。虽然目前学界对CKIP-1介导的自噬通路与骨质疏松症关系的认识已经取得了一定的进展,但还不能完全解释清楚自噬对骨质疏松症的调控机制。相信随着科学技术的发展及学界对自噬研究的不断深入,上述问题一定能被解释清楚,从而能更好地利用与自噬有关的调控体系来防治骨质疏松症。
猜你喜欢
中国骨质疏松杂志(2024年2期)2024-03-19 09:30:14
中国骨质疏松杂志(2021年9期)2021-10-08 10:07:40
天津医科大学学报(2021年3期)2021-07-21 09:03:46
世界科学技术-中医药现代化(2021年12期)2021-04-19 12:31:40
中国临床医学(2019年3期)2019-01-04 09:12:32
安徽医科大学学报(2016年12期)2017-01-15 14:21:53
中国民族医药杂志(2016年6期)2016-05-09 08:52:52
中国医药生物技术(2015年4期)2015-12-26 08:26:36
中国医科大学学报(2015年10期)2015-03-01 02:09:58
中国医学科学院学报(2013年6期)2013-03-11 20:26:04
