
大肠杆菌中吡咯喹啉醌合成途径的构建
2022-09-08 02:55:02杨蒙雅张春月伊进行王怡明卓明洋谢希贤
食品与生物技术学报 2022年8期
杨蒙雅, 张春月, 伊进行, 王怡明, 卓明洋, 马 倩, 谢希贤
(天津科技大学 生物工程学院,天津 300457)
吡咯喹啉醌(Pyrroloquinoline quinone,PQQ),4,5-二羰基-1-吡咯-2,3-f-喹啉-2,7,9-三羧酸,具有抗氧化[1]、维持线粒体功能[2]、介导电子传递等作用,在食品、农业[3]、医疗保健[4]、经济动物养殖[5-6]、传感器制造[7-8]等行业应用广泛,市场前景广阔。目前,PQQ的生产方法主要为化学合成法、提取法和生物发酵法。早在1993年,Martin等人[9]就已经能够通过9步反应将PQQ的产量提升至千克。目前PQQ的生产仍主要依靠化学法合成,但是利用化学法合成PQQ存在步骤多、得率低[10]、对环境污染较大、反应能耗大等问题;提取法主要是从天然产物中提取,但是天然产品中PQQ含量极低,一般在ng/g或ng/mL的范围内[11],且成本较高、利润较低。虽然目前有少量以纳豆为原料经提取法生产的PQQ,但是相关文献报道较少;目前生物发酵法在一些高附加值产品的生产中已经有了成功应用。发酵法合成PQQ能够减少对环境的污染,具有生产过程安全、能耗低、利润大的优势,更加符合产业发展前景。
PQQ生产菌株主要是一些革兰氏阴性菌。其生物合成过程主要由pqq基因簇编码的多个蛋白质催化,不同来源的pqq基因簇存在一些差异,如肺炎克雷伯氏菌[12](Klebsiella pneumoniae)含有一个单独的pqqABCDEF基因簇,而氧化葡萄糖酸杆菌[13](Gluconobacter oxydans) 和 醋 酸 钙 不 动 杆 菌[14](Acinetobacter calcoaceticus)均 含 有 一 个 单 独 的pqqABCDE基因簇,但是还有一些菌株的PQQ合成较为复杂,如Methylovorus sp.MP688[15],除了含有一个单独的pqqABCDE和pqqFG基因簇外,还含有4个单独的pqqA基因;Hyphomicrobium denitrificans FJNU-R8[16]含有3个独立的pqqA基因,一个pqqADE基因簇和一个pqqABCDE基因簇。目前研究的比较多的是氧化葡萄糖酸杆菌,相较其他来源的pqq基因,除pqqA与pqqB之间有一个较短的保守序列外,其余的pqqB、pqqC、pqqD、pqqE之间紧密相连,代谢途径清晰,便于研究。在G.oxydans中,pqqA的作用在于生成一段含有Glu和Tyr的多肽,PqqD则是作为分子伴侣与PqqA形成一定的空间结构,保障PqqA能够在PqqE的作用下完成谷氨酸和酪氨酸残基的连接,PqqC进行PQQ合成最后一步的催化作用,PqqB则与PQQ中间产物在细胞周质空间当中的转运有关[17]。除pqqABCDE基因簇外,氧化葡糖酸杆菌中的tldD基因也与PQQ生物合成有关,可能与其他PQQ合成细菌中的pqqF基因具有类似的功能[18]。
PQQ天然生产菌株都能够产生利用PQQ的蛋白质,与这些菌株类似,大肠杆菌本身含有的D-葡萄糖脱氢酶(GDH)也以PQQ为辅酶,在合成全酶之后能够氧化葡萄糖为葡萄糖酸,由于大肠杆菌自身缺少PQQ合成通路[19],只能依靠摄取外源PQQ使醌蛋白表达功能[20],但是在外源引入pqq基因簇后能够合成PQQ[21],这使得工程化大肠杆菌高产PQQ成为可能。
如表1所示,目前PQQ高产菌株的选育主要集中在菌种筛选和分子改造两个方向,其中诱变筛选[22]、定向进化[23]的效果相比基因改造更加明显,但是定点筛选、定向进化存在周期长、工作量大、遗传背景不清晰、筛选难度大、后续改造困难等一系列问题;以模式微生物为底盘进行分子改造具有周期短、遗传背景清晰、能够精准改造的优点,其中以大肠杆菌为底盘生物进行改造不仅在代谢合成内源性化合物领域具有强大的工业潜力,其在重构异源合成途径代谢生产化学品领域也具有极大的应用前景。
表1总结了部分前人的研究结果,目前对于pqq基因簇的过表达实验研究较少,形式单一,一般是以质粒为表达载体进行单个基因的过表达,虽然改造周期短,效果明显,但是质粒和基因组上过表达的结果有一定差异。除此之外,虽然此前的研究从不同的角度进行了尝试,但是在大肠杆菌中异源表达pqq基因簇还没有系统的研究。作者利用CRISPR/Cas9技 术 将G.oxydans 621H来 源 的pqqABCDE基因簇以及相关的基因在大肠杆菌基因组上进行构建与优化,构建的菌株72 h摇瓶发酵最高产量达到86.3 mg/L,是目前以大肠杆菌为底盘生物进行基因组编辑生产PQQ的最高产量。已报道的以大肠杆菌为底盘生物进行改造的PQQ工程菌最高产量为王朝绚[24]构建的以质粒为载体的PQQ生产菌,产量达到119.8 mg/L。与其相比,本研究的基因组改造表达更稳定,能够为后续以大肠杆菌为底盘生物改造生产PQQ及相关代谢产物提供思路。
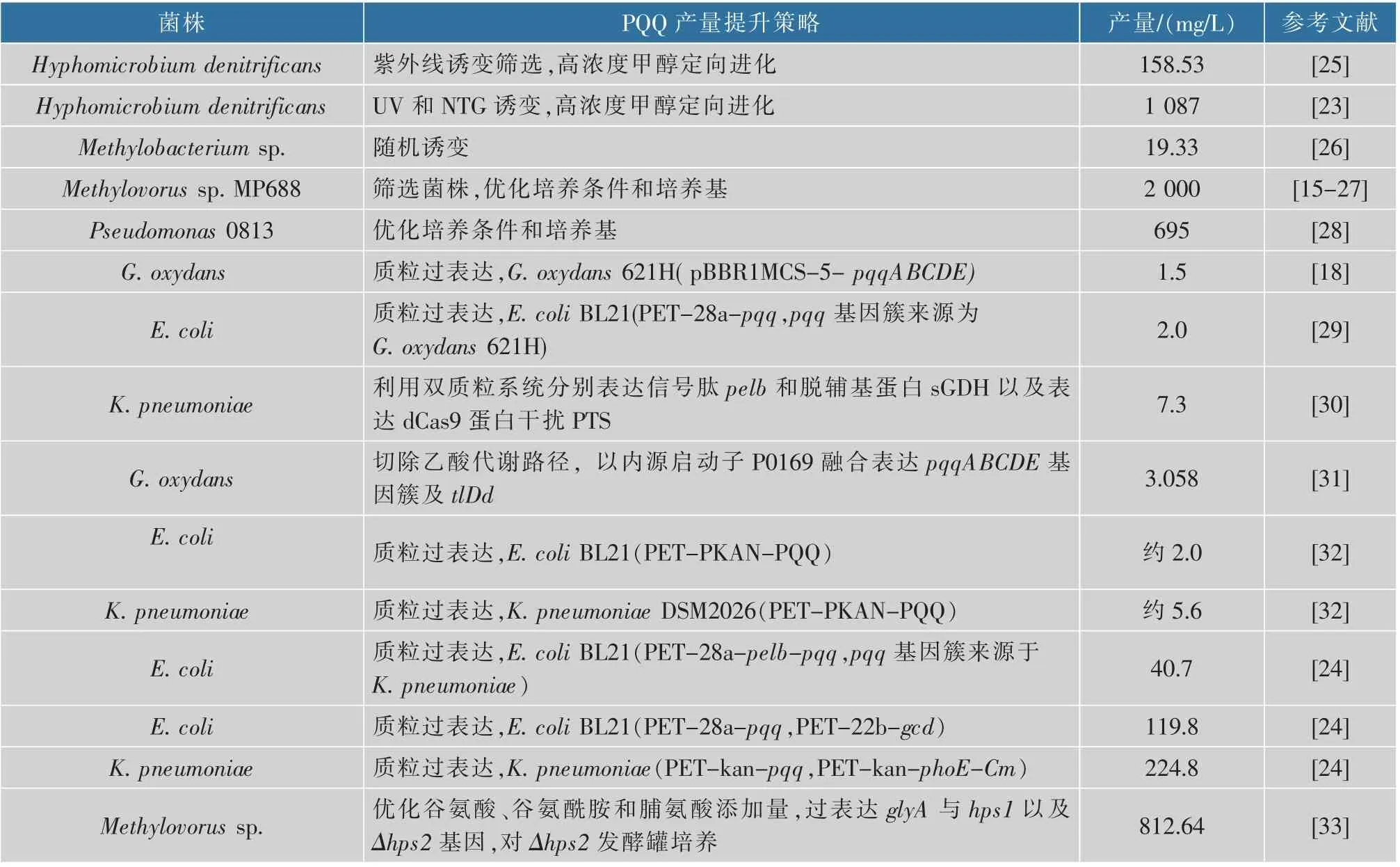
表1 不同微生物中PQQ提升策略及产量Table 1 Strategies and yields of PQQ production improvement in different microorganisms
1 材料与方法
1.1 菌株和质粒
本研究所用的菌株与质粒信息见表2,基因克隆与基因组编辑所用的引物信息见表3,利用E.coliDH5α进行常规的基因克隆工作。以作者所在实验室 构 建 的E.coli W3110、△lacIZ::PxylF-T7RNAP、mlc::mlc*即PQQ-0菌株为出发菌株;限制性内切酶、STAR HS DNA聚合酶、T4 DNA连接酶:均购自TaKaRa;引物、G.oxydans 621H来源的pqq基因簇及tlDd基因:均由苏州金唯智科技有限公司合成;PQQ标品:购自大连美仑生物技术有限公司;KOH和NBT(氯化硝基四氮唑蓝):购自aladdin。

表2 菌株信息Table 2 Strains used in this study

表3 本研究所用引物Table 3 Primers used in this study

续表2
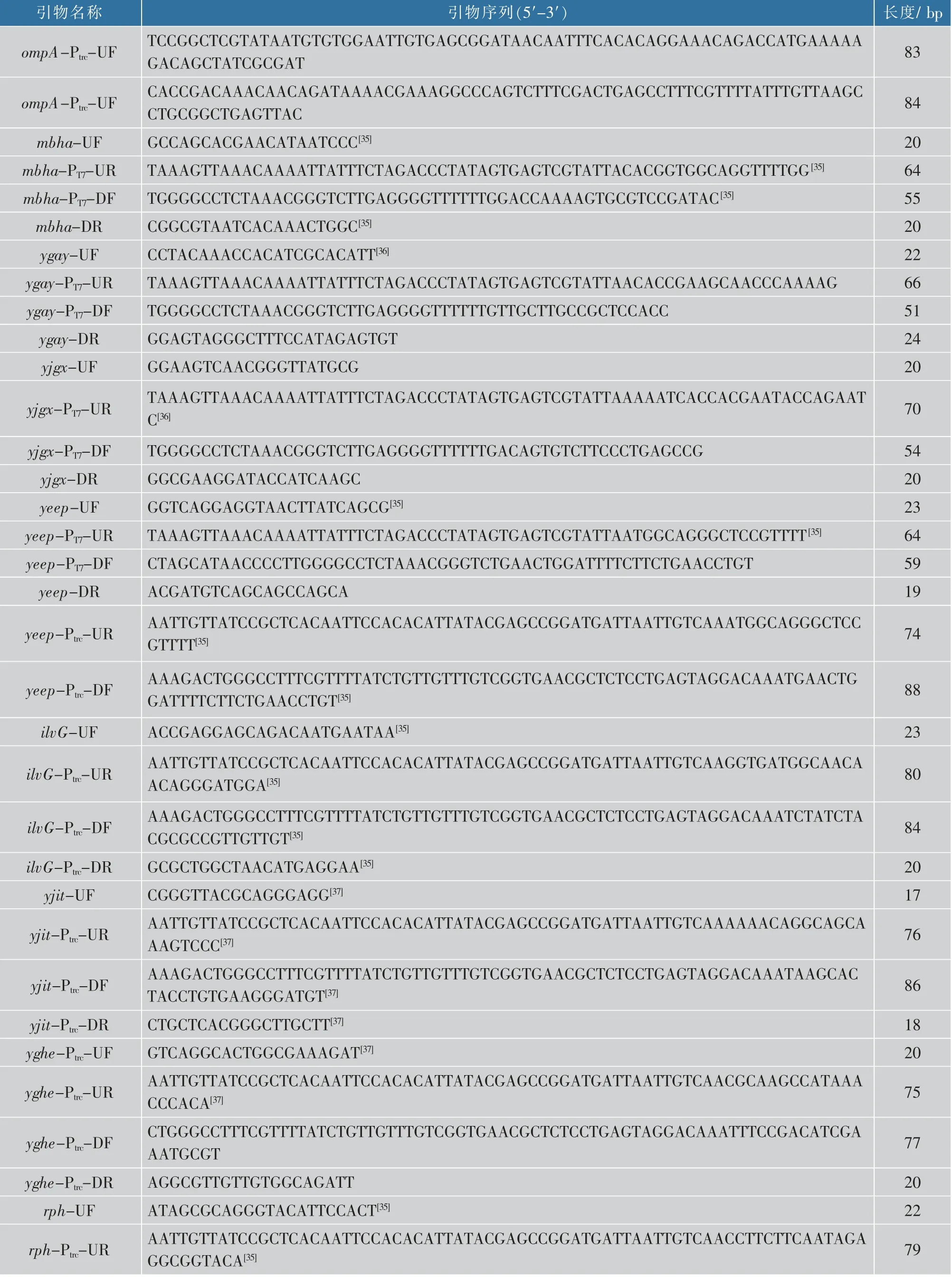
续表3

续表3
1.2 大肠杆菌CRISPR/Cas9基因编辑
1.2.1 重组DNA片段构建根据已知的目的基因序列,利用Primer Primier 5进行引物设计,通过PCR获得目的基因片段。在待敲除或待整合位点分别设计引物,通过PCR扩增500 bp左右的上下游同源臂。进行基因整合时,对上游同源臂、目的基因、下游同源臂进行重叠PCR,获得一段完整的重组DNA片段。进行基因敲除时,对上游同源臂和下游同源臂进行重叠PCR。
1.2.2 pGRB质粒的构建利用CRISPR RGEN Tools(http://www.rgenome.net/cas-designer/)选 择 待整合/敲除位点附近的PAM序列(5′-NGG-3′),并通过合成线性化引物将相应的20 bp的gRNA序列引入单链DNA中,之后通过PCR退火形成双链片段。将双链片段与线性化pGRB载体按照同源重组连接反应 体系 (4 μL 5×CEⅡBuffer,50~200 ng pGRB载体,50~200 ng双链目的片段,2 μL Exnase®Ⅱ重组酶,20 μL ddH2O),化学法转化至E.coli DH5α感受态中,筛选阳性克隆并提取质粒。
1.2.3 基因整合与敲除过程根据文献[34]报道的CRISPR/Cas9基因编辑技术进行E.coli基因组DNA的整合和敲除。操作体系由目标重组DNA片段、提供gRNA片段的pGRB质粒和提供Cas9蛋白及RED重组酶的pREDCas9质粒构成。具体方法为:先将pREDCas9质粒转化至待改造的E.coli感受态细胞中,然后将上述重组pGRB质粒和重组DNA片段电转至含有pREDCas9质粒的感受态细胞中,电转后加入SOC复苏液于32℃培养2 h,取100 μL菌液涂布于AmpR和SpeR抗性的培养皿,32℃培养,筛选阳性克隆。将筛选到的阳性菌株在含0.2 g/dL阿拉伯糖的LB培养基中培养,诱导pREDCas9中的pGRB消除系统,对pGRB质粒进行消除,筛选在奇霉素抗性平板生长但在氨苄霉素抗性平板不生长的阳性克隆,即为丢失了pGRB质粒的克隆。利用pREDCas9的温敏特性,可以在42℃培养过夜消除,筛选在无抗平板生长而在SpeR平板不生长的单克隆菌株,即为相应改造的无质粒工程菌株[34]。
1.3 摇瓶发酵
首先在5 mL LB液体培养基(蛋白胨10 g/L,NaCl 10 g/L,酵母粉5 g/L)中进行菌种活化,其次在固体斜面培养基(蛋白胨10 g/L,酵母粉5 g/L,NaCl 10 g/L,牛肉膏10 g/L,蔗糖1 g/L,琼脂粉20 g/L)进行菌种二代活化,置于37℃培养箱中培养12 h后转移至新的固体斜面进一步培养12 h。用接种环刮取菌体,接种于500 mL三角瓶中(含有30 mL培养基),用九层纱布封口,在37℃、220 r/min条件下培养10~12 h至OD600为8.0左右。种子培养基成分为:葡萄糖20 g/L,蛋白胨10 g/L,NaCl 10 g/L,酵母粉5 g/L,体积分数2%苯酚红溶液,消泡剂1滴,pH 7.0~7.2。
将培养好的种子培养液按照10%的接种体积分数接种于30 mL发酵培养基,在37℃、220 r/min条件下培养至发酵结束。发酵培养基成分为:葡萄糖20 g/L,Na2HPO4·12H2O 17.12 g/L,KH2PO43 g/L,NaCl 0.5 g/L,NH4Cl 1 g/L,CaCl26H2O 0.219 g/L,MgSO4·7H2O 0.492 g/L,木糖10 g/L,2%苯酚红溶液,pH 7.0,消泡剂1滴。在发酵过程中,根据苯酚红指示剂的颜色变化,及时以氨水调整pH至7.0左右,以60 g/dL的葡萄糖溶液补充碳源。
1.4 PQQ检测方法
1.4.1 几种检测方法的比较目前检测PQQ的方法有高效液相色谱法、液质联用法、酶法检测、质谱检测、非酶法检测等。PQQ极易溶于水,且携带3个羧基,显酸性,液相检测法在15.625~500 mg/L范围内线性关系良好,但是检测时易拖尾且峰宽,导致检测结果不准确[39-40]。比高效液相色谱法更精确的检测方法如液质联用[41]等检测限也都远高于实际产量,且检测前需要对发酵液进行预处理,由于在预处理过程中可能导致一部分PQQ损失,造成结果不准确。酶法检测比较灵敏,能够检测到20×10-12g的PQQ[42],但是由于PQQ易与亲核的氨基酸和蛋白质反应[43],因此目前在大肠杆菌中构建PQQ代谢通路选择的培养基均为细菌基本培养基,该培养基中仅有的碳源为葡萄糖,而发酵液中含有Mg2+、Ca2+,酶法检测的原理是预先混合PQQ和纯化后的D-GDH(或粗酶液)反应一段时间后形成有活性的DGDH,随后加入反应液[44](含DCIP、PMS、葡萄糖、MgSO4的混合溶液),形成了氧化还原反应完整的反应链,通过计算DCIP的褪色速率与标准品比较可以计算得到发酵液中PQQ的浓度。但是发酵液中浓度不等的残留葡萄糖会对检测结果产生较大的影响,尤其对于补料分批培养。非酶法检测PQQ[45]应用广泛,主要利用PQQ在碱性条件下氧化甘氨酸,将电子传递给NBT,使得NBT生成蓝紫色的甲臜类化合物,该物质在OD530有最大吸光度,非酶法检测不仅能够同时处理多个样品,检测限可以低至3.906 ng/mL,而且发酵液中的残糖对其无影响[24,28,32-51]。基于以上比较,本实验选择的检测方法为非酶法。
1.4.2 PQQ的非酶法检测将待测样品于13 000 r/min离心3 min,取上清液。依次在96孔板中加入180 μL Gly-KOH溶液(2 mol/L Glycine,用KOH将pH调至10),80 μL PB溶液(NaH2PO4·2H2O 1.911 g/L,Na2HPO4·12H2O 2.776 g/L),20 μL样品,20 μL NBT母液(NBT固体2.943 g/L,用20 mmoL PB溶液溶解,现配现用),混匀后于30℃避光孵育1 h,用酶标仪检测OD530条件下的吸光度。
1.5 菌体量的计算
使用分光光度计测量菌液稀释后的OD600,并且对烘干后的菌体进行称重,得到细胞干质量与OD600之间的对应关系。
DCW(g/L)=0.382×OD600×稀释倍数。
2 结果与讨论
2.1 过表达pqqA和pqqB对PQQ产量及生物量的影响
在PQQ的合成中,PqqA作为其前体物质,在分子伴侣PqqD的保护下经过PqqE的作用完成谷氨酸和酪氨酸残基的连接,PqqB负责中间产物在周质空间当中的转运,PqqC负责合成最后的催化,TlDd可能起到切割作用。PqqA作为前体物提升其表达量能够促进PQQ的产生。以作者所在实验室构建的E.coli W3110,△lacIZ::PxylF-T7RNAP,mlc::mlc*即PQQ-0为出发菌,该菌株基因组上已经整合了T7RNA聚合酶,可以通过添加木糖控制T7RNA聚合酶的表达。由于基因距离启动子的位置不同会导致表达量不同,柯崇榕等[16]以双质粒系统分别过表达了脱氮生丝微菌来源的pqqA和pqqBCDEF,与单质粒系统相比,产量有所提升。杨雪鹏等[29]比较了G.oxydans 621H来源的pqq基因簇在不同质粒中的表达情况,发现以PET-28a为表达载体产量最高,能够达到约2 mg/L,说明PT7启动子也适合于PQQ的合成。因此,在PQQ-0的基础上,我们在mbha和ygay这两个假基因位点以PT7为启动子整合了pqqA和pqqBCDE基因簇,打通了PQQ合成通路,构建了PQQ-1菌株。
G.oxydans 621H中pqqABCDE共用一个启动子,Li等[46]的研究表明,pqqA的表达强度远高于其余几个pqq基因,这也符合PqqA作为PQQ合成前体物的特征。李盼盼[47]的研究也表明,外源添加PqqA就能够提升PQQ产量。PQQ的结构中有谷氨酸和酪氨酸,虽然谷氨酸和酪氨酸不是其直接的前体物,但已有实验证明了外源添加谷氨酸和酪氨酸能够提升PQQ的产量[31-32]。增强pqqA的表达可以为PQQ的合成提供直接前体物,因此,本研究在菌株PQQ-1中利用PT7启动子强化了pqqA的表达,得到了PQQ-2菌株,摇瓶发酵72 h的结果见图1。PQQ-1产 量 为62.6 mg/L,强 化 了PqqA合 成 的PQQ-2菌 株 产 量 为67.8 mg/L,较PQQ-1提 升8.3%,且生物量提升4.3%。可见,强化pqqA的表达对PQQ的合成与菌体生长均有促进作用。

图1 过表达pqqA和pqqB对PQQ合成及菌体量的影响Fig.1 Effects of over-expression of pqqA and pqqB on PQQ synthesis and biomass
在PQQ-2的基础上,进一步强化pqqB的表达,分别以Ptrc和PT7为启动子,在PQQ-2中同一假基因位点过表达了pqqB,构建了菌株PQQ-3和PQQ-4。PQQ-3菌株与PQQ-4菌株72 h PQQ产量较PQQ-2均上升,产量分别为68.8 mg/L和82.3 mg/L。从生物量的角度考虑,PQQ-3的生物量较PQQ-2下降了3.6%,下降的幅度较小,而PQQ-4的生物量较PQQ-2下降了9.3%,也低于PQQ-1的菌体量。PQQ-4在摇管活化培养中也出现了菌体早衰的现象,结合后续改造的需要考虑,我们选择了PQQ-3作为下一步改造的出发菌。PQQ-3和PQQ-4的产量和菌体量的差异可能是由于PT7的启动强度较Ptrc更强,PQQ-4在表达更多PqqB蛋白的同时对菌体的生长影响更大。Wang等[48]在G.oxydans WSH003中以pqqA的启动子和tufB启动子分别过表达pqq单个基因时也印证了强启动子能够更大程度促进PQQ的生产。
2.2 过表达pqqC、pqqD及pqqE对PQQ产 量及菌体量的影响
选择筛选出的PQQ-3作为出发菌,探索过表达pqqC、pqqD、pqqE对产量提升以及对菌体生长的影响。由于PT7启动子过强,在PQQ-4的培养和发酵过程中发现对菌体生长影响较大,因此后续改造均选择Ptrc启动子进行相关基因表达控制。分别在PQQ-3菌株中引入Ptrc控制的pqqC、pqqD、pqqE基因,得到PQQ-5、PQQ-6、PQQ-7菌株;在PQQ-5菌株中引入Ptrc控制的pqqC,构建了PQQ-8;在PQQ-6中引入Ptrc控制的pqqD,得到PQQ-9;在PQQ-7中引入Ptrc控制的pqqE,得到PQQ-10,发酵结果见图2。改造后只有PQQ-7产量较PQQ-3有所上升,72 h产量能够达到74.8 mg/L,较改造前上升了8.7%,其余的菌株PQQ产量在改造后均出现了下降。

图2 过表达pqqC、pqqD及pqqE对PQQ产量及菌体量的影响Fig.2 Effects of over-expression of pqqC,pqqD and pqqE on PQQ yield and biomass
本研究的结果表明,pqqA、pqqB和pqqE在大肠杆菌基因组上的过表达对PQQ产量的提升有正向作用,且在pqqA的过表达中强启动子PT7的效果优于Ptrc。但是对于pqqC和pqqD的表达后出现了产量下降,推测原因可能是在PQQ的合成过程中,PqqC蛋白行使的是PQQ生成最后一步的催化功能,PqqD蛋白作为pqqA的分子伴侣,其参与的反应可能不是PQQ合成的限速步骤,因此在pqqC和pqqD过表达以后,降低了其他关键酶的表达量,导致PQQ-5和PQQ-6较PQQ-3产量下降,因此选择PQQ-7作为下一步改造的出发菌株。
2.3 过表达不同来源的tlDd基因对PQQ产量及生物量的影响
G.oxydans 621H中对PQQ合成有影响的除了pqqABCDE以外,还有距离该基因簇较远的tlDd基因,该基因编码TlDd蛋白,TlDd蛋白可能执行PqqF的剪切功能[18]。对G.oxydans 621H中tlDd进行密码子优化后,序列比对显示,G.oxydans 621H与E.coli K-12 W3110中的tlDd基因同源性高达56%。大肠杆菌自身含有的tlDd可能在PQQ合成中发挥作用,这也是只在E.coli中过表达pqqABCDE基因簇就能合成PQQ的原因之一。为了比较两个来源的tlDd基因过表达对PQQ产量的影响,我们在PQQ-7菌株的基础上构建了4个菌株,均选择对生长影响较小的Ptrc启动子进行过表达。
为了探究不同来源的tlDd基因对PQQ产量提升的影响,在PQQ-7菌株上以Ptrc启动子过表达E.coli来源的tlDd基因,分别得到了tlDd双拷贝菌株PQQ-11和三拷贝菌株PQQ-13。在PQQ-7菌株上以Ptrc启动子过表达G.oxydans 621H来源的tlDd基因,分别得到了tlDd双拷贝菌株PQQ-12和三拷贝菌株PQQ-14。发酵72 h后产量和菌体量见图3。在不同批次的发酵中,由于摇瓶发酵过程中补氨和补糖的差异,导致PQQ-7产量和菌体量的差异。PQQ-11、PQQ-12、PQQ-13、PQQ-14这4株菌较PQQ-7产量均有所上升,说明本研究选择的E.coli和G.oxydans两种不同来源的tlDd基因对于PQQ的产量提升都有促进作用,而PQQ-11较PQQ-12的产量更高,原因可能为大肠杆菌对自身来源的基因适配性更好。对于同一来源的tlDd基因过表达,PQQ-11较PQQ-7产量和菌体量分别提高25.1%和5.2%;PQQ-12较PQQ-7产量提高9.8%,菌体量几乎一样,但是两个参数的提升量均低于PQQ-11,说明在PQQ-11中进一步强化表达tlDd基因会对菌体量和产量均产生不利影响。综合PQQ产量和生物量两个参数的结果,选择PQQ-11作为下一步改造的出发菌株。
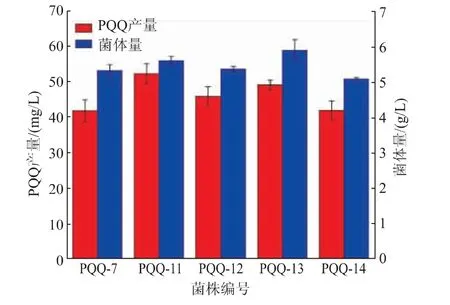
图3 过表达不同来源的tlDd基因对PQQ产量及生物量的影响Fig.3 Effects of over-expression of tlDd from different source on PQQ yield and biomass
2.4 敲除iscR、过表达gcd和ompA对PQQ产量及菌体量的影响
大肠杆菌中存在铁-硫簇抑制[49],柯崇榕的研究表明,在培养基中加入Fe2+或敲除大肠杆菌中Fe-S簇的转录抑制子iscR均能提高PqqE的表达量,但是通过添加Fe2+的方法需要实验确定添加量,过量反而会抑制PQQ的生产[16],因此尝试通过敲除iscR解除铁-硫簇抑制。在PQQ-11菌株中敲除iscR基因,获得PQQ-15菌株,PQQ产量由52.4 mg/L提升为69.2 mg/L,提升了32.1%,菌体量由5.6 g/L提升为6.4 g/L,提升了14.3%,见图4。iscR的敲除在本研究中对PQQ提升的作用最大,表明E.coli中可能还存在一些内源基因对PQQ的合成有抑制作用。

图4 敲除iscR、过表达gcd和ompA对PQQ产量及生物量的影响Fig.4 Effects of knockout of iscR,and over-expression of gcd and ompA on PQQ yield and biomass
目前报道的PQQ的高产菌株仍然以甲基营养菌居多,经过分析发现,这类菌株的细胞膜上有多种能够利用PQQ的蛋白质[50]。由于GDH的合成并不依赖于PQQ,但是PQQ的合成却在GDH合成之后才开始[43]。韩增叶[51]的研究表明,在大肠杆菌中引入外源PQQ合成途径后,在高浓度葡萄糖的刺激下,GDH的表达量有明显提升。由于GDH本身是脱辅基蛋白质,只有在和PQQ以及Mg2+或Ca2+形成全酶后才有活性。随着PQQ产量的增加,势必要提升离子的浓度来适配高产量的PQQ,因此优化Mg2+添加量[51]后能够通过提升GDH的表达量间接促进PQQ的生产。在PQQ-15菌株的基础上,以Ptrc启动子过表达E.coli K-12 W3110自身来源的gcd基因,得到了菌株PQQ-16,与PQQ-15相比,PQQ-16产量由69.2 mg/L提升至86.2 mg/L,提升了24.6%,菌体量也由改造前的6.4 g/L提升至6.6 g/L。
ompA是大肠杆菌孔蛋白基因,文献[24]在肺炎克雷伯氏菌中过表达ompA,发现促进了胞内PQQ向胞外转运[24]。因此在PQQ-16的菌株基础上以Ptrc强启动子过表达了ompA孔蛋白基因,试图将胞内产生的PQQ及时转运出胞外,避免其在胞内的消耗,解除可能存在的反馈抑制。结果表明,与PQQ-16相比,PQQ-17产量略有上升,为86.3 mg/L,菌体量有一定上升,为7.0 g/L。结果表明,以强启动子Ptrc过表达E.coli自身来源的ompA孔蛋白基因能够有限提升胞外PQQ浓度,但是产量提升幅度非常小,说明在PQQ-16菌株中,PQQ从胞内向胞外的转运并没有受到孔蛋白OmpA表达量的限制。
作者系统研究了异源表达G.oxydans 621H操纵子pqqABCDE以及优化底盘相关基因对PQQ合成的影响,获得了一株能够高效合成PQQ的工程菌,摇瓶发酵72 h产量达到86.3 mg/L。
3 结语
作者成功构建了PQQ生产工程菌,为了进一步提升PQQ的产量,后续改造思路为对发酵培养基及发酵条件进行优化[24,47-52],如培养基中的酪氨酸和谷氨酸添加量、金属离子添加量、发酵过程中的溶氧等;阻断部分非必要的碳源代谢降低副产物乙酸的产生[31]。
猜你喜欢
当代水产(2022年1期)2022-04-26 14:35:38
黑龙江大学自然科学学报(2021年4期)2021-11-19 07:05:02
西北农林科技大学学报(自然科学版)(2019年8期)2019-07-17 02:43:32
食品科学(2018年10期)2018-05-23 01:27:28
中国调味品(2017年2期)2017-03-20 16:18:21
现代检验医学杂志(2016年3期)2016-11-15 01:59:48
西南医科大学学报(2015年1期)2015-08-22 13:01:46
中国当代医药(2015年9期)2015-03-01 02:01:59
遗传(2015年5期)2015-02-04 03:06:55
西南军医(2015年6期)2015-01-23 01:25:50
