
RuMoSn@MOF-801纳米催化剂的制备及其催化氨硼烷制氢性能研究
2022-09-07 03:45:40徐晨薇童倩倩莫晗陈驰赵培森王峥
湖北大学学报(自然科学版) 2022年5期
徐晨薇,童倩倩,莫晗,陈驰,赵培森,王峥
(有机化工新材料湖北省协同创新中心,有机功能分子合成与应用教育部重点实验室,湖北大学化学化工学院,湖北 武汉430062)
0 引言
21世纪的挑战之一是开发可再生绿色能源,以取代当今的化石能源.我国承诺2030年前,二氧化碳的排放不再增长,达到峰值之后逐步降低.碳排放达峰的本质是实现绿色低碳转型.氢可代替碳氢化合物,用于储能,也可作为燃料使用,被视为21世纪最具有发展潜力的清洁能源之一[1].然而,氢气的高效储存和可控释放仍然是氢能源可持续发展的制约因素. 除高压储氢外,氢可以通过化学方式储存在其他材料中,如金属氢化物、水合肼、硼氢化物和氨硼烷.其中氨硼烷(NH3BH3,简写为AB)是化学储氢的典型代表[2].AB储氢密度为196 g H2·kg-1,可通过水解[3-4]、热分解和在非水溶剂中催化脱氢实现氢的释放[5].室温下,1 mol AB水解催化可释放3 mol H2. AB水解制氢可以通过催化剂进行可控放氢,具有反应条件温和、不产生CO等优点[6].
研究表明,Pt、Pd、Ru和Rh等贵金属对AB催化制氢具有良好活性[7].但是,这些贵金属成本高,资源有限,限制了其规模化应用. 在催化 AB制氢的贵金属催化剂中,Ru成本相对较低,通过降低贵金属的用量,最大限度地提高贵金属的利用效率,制备出低成本、高效催化剂,对氨硼烷水解制氢至关重要[8].近年来,三金属纳米催化体系在催化氨硼烷水解制氢中,表现出较高活性,通过掺杂非贵金属,降低成本,且在室温AB制氢中表现出更好的催化性能,受到广泛的关注[9].然而,在催化剂的制备过程中,金属纳米粒子很容易在溶液中团聚,且在空气中易氧化,因此需要寻找合适的载体来解决这个难题.

1 实验方法
1.1 催化剂的制备
1.1.1 MOF-801的制备[17-18]称取ZrOCl2·8H2O(1.6 g, 5 mmol, 1 eq)和反丁烯二酸(0.58 g, 5 mmol, 1 eq)溶于27 mL DMF-甲酸 (体积比为20∶7)混合溶液中.完全溶解后,将澄清溶液转移到50 mL反应釜,在130 ℃条件下反应6 h.反应结束后,冷却至室温.将所得产物离心分离,用DMF和甲醇交替洗涤3次,去除残留反应物,50 ℃真空干燥14 h.MOF-801的合成过程如图1所示.
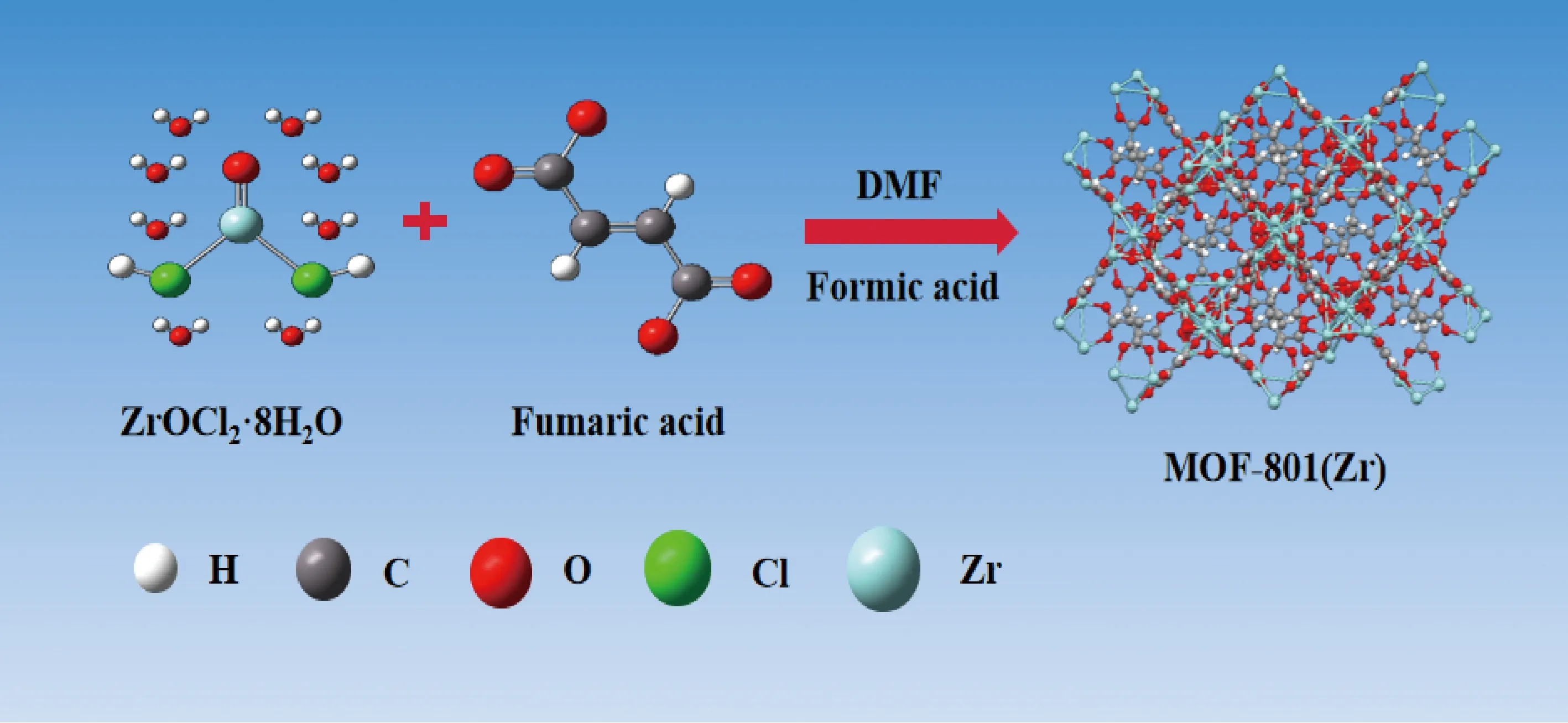
图1 MOF-801的合成过程示意图
1.1.2 Ru1Mo0.5Sn0.1@MOF-801的制备 将50 mg MOF-801均匀分散在30 mL乙醇中,超声20 min,用移液枪分别将3.5 mL RuCl3(0.01 mol/L),0.175 mL Na2MoO4(0.1 mol/L)和0.035 mL SnCl4(0.1 mol/L)加到MOF-801悬浊液中(A),磁力搅拌5 h,将50 mg NaBH4溶于10 mL去离子水,逐滴加到A中,使Ru(III)、Mo(Ⅵ)和Sn(Ⅳ)还原,继续搅拌3 h,抽滤, 50 ℃真空干燥12 h,得到Ru1Mo0.5Sn0.1@MOF-801.采用上述相似步骤,分别制得Ru@MOF-801、Ru1Mo0.5@MOF-801、Mo0.5Sn0.1@MOF-801以及金属Mo/Sn摩尔比不同的三金属催化剂RuMoxSny@MOF-801 (x=0.1,0.3,0.5,0.7,1.0;y=0.05,0.1,0.3,0.5).
1.2 主要试剂及仪器
1.2.1 试剂 实验所用试剂均为分析纯.八水氧氯化锆(≥ 99.0%)、反丁烯二酸(≥ 99.0%)、N,N-二甲基甲酰胺(≥ 99.8%)、甲酸(≥ 99.0%)、无水甲醇(≥ 99.8%)、钼酸钠(≥ 99.9%)、二水四氯化锡(≥ 99.9%)和硼氢化钠(≥ 98.0%)均购于国药集团化学试剂有限公司;氨硼烷(≥ 97.0%)购于郑州阿尔法化工有限公司;三氯化钌水合物(≥ 97.0%)购于阿拉丁化学试剂有限公司.
1.2.2 仪器 D8-Advance X 线粉末衍射仪(德国Bruker 公司),Cu 靶Kα (λ=0.154 178 nm)为射线源,测试温度为室温,工作电压为40 kV,工作电流为40 mA,扫描范围为5°~80°,扫描速度为5°/min.高分辨透射电子显微镜(HRTEM,FEI-Tecnai F20,美国FEI公司);Sigma 500 型场发射扫描电镜(德国ZEISS 公司);ESCALAB 250Xi 型光电子能谱仪(XPS,美国Thermo 公司);电感耦合等离子体原子发射光谱(ICP-AES, IRIS Intrepid IIXSP,美国Thermo 公司);Micromeritics ASAP 2020 比表面积分析仪(美国麦克公司);Impact 420 型FT-IR 红外光谱分析仪(美国Nicolet 公司).
1.3 催化活性测试催化实验在连接有恒压漏斗和气体监测装置的双颈圆底烧瓶中进行,圆底烧瓶中预先加入10.0 mg催化剂,恒压漏斗中盛有 10 mL氨硼烷水溶液(18.5 mg AB).水浴恒温 25 ℃条件下,一次性加入氨硼烷水溶液后,开始计时,每间隔 30 s 记录一次气体体积.采用同样的方法,分别测定AB在25、30、35、40 ℃条件下的水解速率,计算反应活化能.
2 结果与讨论
2.1 XRD和FT-IR分析
2.1.1 XRD分析 图2为制备得到的载体MOF-801和负载后的催化剂XRD谱图, MOF-801载体的XRD特征衍射峰位于8.52°、9.86°和25.75°,分别对应MOF-801的(111)、(002)和(244)面,与MOF-801文献模拟谱图吻合[19],表明制备得到的MOF-801与文献报道的MOF-801结构相同.此外,制备得到的MOF-801XRD谱图无杂峰,说明制备得到的MOF-801具有较高的相纯度.负载金属后,5组催化剂的衍射峰虽有不同程度的减弱,但衍射峰的形状和位置与MOF-801基本一致,表明催化剂在制备和催化过程中结构保持不变,催化剂表现出良好的稳定性.图2中没有观察到Ru、Mo、Sn明显的特征衍射峰,这可能与金属的负载量较低,或金属是以非晶相存在有关[20].
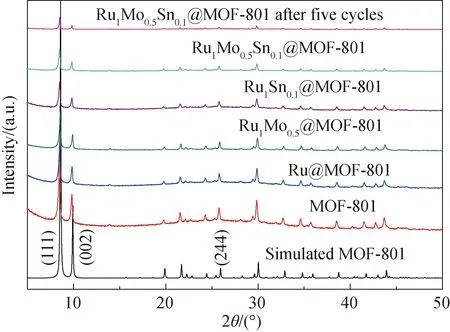
图2 不同催化剂的XRD图谱
2.1.2 FI-IR分析 图3为载体MOF-801、催化剂Ru1Mo0.5Sn0.1@MOF-801和5次催化循环后的Ru1Mo0.5Sn0.1@MOF-801的红外光谱.制备得到的MOF-801与文献报道的MOF-801数据一致[21-22]. 1 402 cm-1附近的吸收峰归因于羧基O—C—O的对称拉伸,1 646 cm-1和1 578 cm-1处的谱带归因于该基团的不对称拉伸.794 cm-1,984 cm-1和1 212 cm-1的吸收峰可归为C—H振动.490 cm-1和660 cm-1处的两个峰归因于Zr—(OC)的不对称拉伸和Zr6(OH)4O4团簇的振动.3种样品的红外光谱图较为相似,说明催化剂Ru1Mo0.5Sn0.1@MOF-801经过5次催化循环后,载体MOF-801的骨架结构仍然保持不变,即5次循环实验并没有破坏Ru1Mo0.5Sn0.1@MOF-801骨架的完整性.

图3 不同催化剂的红外光谱图
2.2 形貌和组成分析图4(a)为载体MOF-801 FESEM照片,从图中可以看出,MOF-801形状规则,粒径均匀.图4(b)为Ru1Mo0.5Sn0.1@MOF-801催化剂的HRTEM照片,由图可知,催化剂Ru1Mo0.5Sn0.1@MOF-801材料的晶格条纹间距为0.153 nm和0.180 nm,分别对应ZrO2的(121)和(112)晶面[18].图4(c)为催化剂Ru1Mo0.5Sn0.1@MOF-801的TEM照片,可以看出RuMoSn NPs均匀负载在载体MOF-801上,没有出现团聚现象.图4(d)为Ru1Mo0.5Sn0.1@MOF-801负载金属的粒径分布图,负载的金属粒径主要分布在0.3~2.4 nm,平均粒径为1.35 nm.图4(e)为纯RuMoSn NPs的TEM照片,其粒径如图4(f)所示,分布在42.3~58.4 nm,平均粒径为50.3 nm.可见,金属纳米粒子未分散到载体上时,易发生团聚,金属颗粒尺寸较大,会导致金属颗粒比表面积减小.
图5(a)和5(b)为Ru1Mo0.5Sn0.1@MOF-801的FESEM照片,与图4(a)对照, MOF-801负载金属纳米粒子后,形貌和粒径基本保持不变.图5(c)为EDS谱图,图中显示该催化剂含有Ru、Mo、Sn、Zr、O元素.图5(d)~5(f)为Ru1Mo0.5Sn0.1@MOF-801催化剂的元素分布图,表明Ru、Mo、Sn金属粒子均匀地负载到MOF-801上,且载体是含Zr的金属有机框架.

图5 (a, b) Ru1Mo0.5Sn0.1@MOF-801的FESEM照片、(c) EDS能谱分析和(d~f)相应的元素分布图像
2.3 XPS分析XPS全谱图(图6(a))确认催化剂Ru1Mo0.5Sn0.1@MOF-801含有C、O、Zr、Ru、Mo、Sn 6种元素.由图6 (b)可知,Zr 3 d峰值位于181.9和184.4 eV处,对应Zr(IV)的3d5/2和3d3/2[23].图6 (c)为O 1s的信号峰, O 1s可分为2个峰,峰值位于529.3 eV和531.2 eV 处,分别对应Zr-O和Zr-OH[22].图6(d)为Ru 3p的信号峰,Ru峰值位于463.4和486.3 eV处,分别对应Ru(0)的3p3/2和3p1/2[24].图6 (e)为Mo 3d 信号峰,Mo峰值位于231.8 和235.0 eV处,分别对应Mo(Ⅵ)的3d5/2和3d3/2自旋轨道态[25].图6 (f)为Sn的3 d信号峰,Sn峰值位于486.4 eV和494.8 eV处,分别对应于Sn(0)的Sn 3d5/2和Sn 3d3/2自旋轨道态;Sn峰值位于487.5 eV和495.8 eV,分别对应Sn(IV)的3d5/2和3d3/2自旋轨道态[26].XPS研究结果表明,在Ru1Mo0.5Sn0.1@MOF-801催化剂中,Ru单质仅以零价形式稳定存在,而Mo、Sn 在制备过程中可能发生了一定程度的氧化.
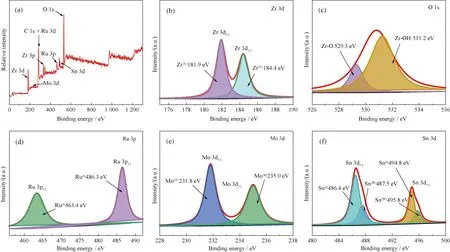
图6 Ru1Mo0.5Sn0.1@MOF-801催化剂的XPS谱图
2.4 ICP-AES分析
2.4.1 ICP-AES测试样品的制备 称取10 mg Ru1Mo0.5Sn0.1@MOF-801催化剂,逐滴滴加3 mL 王水(浓硝酸和浓盐酸体积比为1∶3)使其完全消解,待其完全消解后,取1 mL该溶液,用去离子水稀释至10 mL,重复该操作直至溶液澄清透明,取10 mL 溶液进行ICP-AES测试.
2.4.2 ICP-AES测试过程 采用5个不同浓度(0 mg/L, 0.5 mg/L, 1 mg/L, 10 mg/L, 50 mg/L)的Ru、Mo和Sn标准溶液做标准工作曲线,测定未知溶液浓度,通过计算,实验结果如表1所示.Ru、Mo和Sn的实际负载量(质量分数)分别为2.01%、1.05%和0.18%,原子比为Ru∶Mo∶Sn=1∶0.52∶0.09.此结果与理论加入量Ru∶Mo∶Sn=1∶0.5∶0.1相近,表明浸渍还原法可以成功地将金属纳米粒子还原并定量负载到载体MOF-801上.

表1 RuMoSn@MOF-801催化剂的元素分析
2.5 BET分析对载体MOF-801、催化剂Ru1Mo0.5Sn0.1@MOF-801进行N2吸附BET比表面积和孔结构分析.MOF-801和Ru1Mo0.5Sn0.1@MOF-801的比表面积分别为1 023.78 m2/g,26.28 m2/g;孔体积分别为0.896 cm3/g,0.184 cm3/g.负载后,MOF-801的比表面积和孔体积显著减小,这是由于载体MOF-801的表面及孔洞负载了RuMoSn NPs造成的.
2.6 催化性能分析图7为25 ℃时,不同催化剂催化AB水解产氢速率图.由图7(a)可知,与单金属Ru@MOF-801和双金属RuMo@MOF-801、RuSn@MOF-801、MoSn@MOF-801负载型催化剂相比,三金属RuMoSn@MOF-801表现出更高的催化活性,证实贵金属Ru和非贵金属MoSn纳米颗粒之间存在明显的协同作用,这种协同作用可能是由Ru、Mo、Sn之间的电子效应[9]和几何效应[27]引起的.Ru、Mo、Sn的电负性分别为2.20、2.16、1.80,其中,Ru、Sn金属间的电负性差值较大,它们之间将产生强的电子效应;而Ru、Mo金属的电负性相近且晶体结构相似,在金属合金形成过程中,Mo原子可能会取代少部分Ru原子,保留Ru团簇的主要hcp结构,使Ru、Mo两种金属之间产生几何效应.此外,纯载体MOF-801 对AB的水解反应没有任何活性,且纯RuMoSn NPs的催化活性远低于三金属负载型RuMoSn@MOF-801,这归因于RuMoSn NPs与载体MOF-801之间的双功能作用.由图4(d)、4(f)可知,负载后的RuMoSn NPs尺寸受到MOF-801空间上的限制,尺寸明显减小,由此增大了RuMoSn NPs与AB分子的有效接触面积,促进水解反应活性中间体的形成,提供更多有利于AB水解反应的活性位点,尽管载体MOF-801自身对AB水解产氢没有催化活性,但却能为RuMoSn NPs 提供良好的3D框架.
RuMoSn@MOF-801催化剂对AB水解反应的催化机理可能包含两个步骤[9]:首先,AB与RuMoSn NPs表面相互作用,形成活化的瞬态金属-H,这是水解反应的先决条件;然后,H2O分子对金属-H攻击,释放H2.由于RuMoSn均匀分散在MOF-801的表面和孔洞中,使得金属纳米颗粒与AB的接触面积增加,活化的瞬态金属-H的活性位点数增加.因此,RuMoSn@MOF-801催化AB水解具有高产氢率.
图7(b) 和图7(c)分别是Ru1MoxSny@MOF-801(x=0.1,0.3,0.5,0.7,1.0,y=0.1)和(x=0.5,y=0.05,0.1,0.3,0.5)的AB水解制氢速率图.由图可知,催化反应速率随着Mo/Sn摩尔比的增加先增加再降低,这是因为当反应物浓度到达一定值后,反应物与催化剂的表面结合力过强,导致反应物占据催化剂的有效表面位置而使催化剂中毒[27].结果表明,当Ru1MoxSny@MOF-801中,Ru,Mo和Sn的摩尔比为1.0∶ 0.5∶ 0.1时, Ru1Mo0.5Sn0.1@MOF-801催化剂具有最高的催化活性.
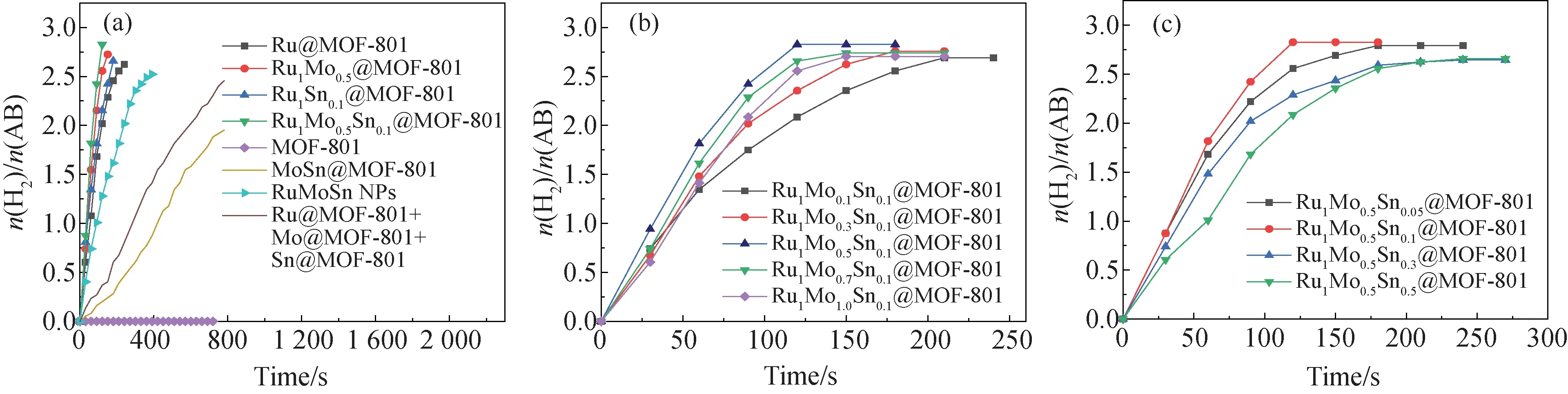
图7 不同催化剂催化AB水解制氢速率图
为了测定Ru1Mo0.5Sn0.1@MOF-801催化 AB 水解反应的活化能(Ea),在25 ~ 40 ℃下测定了催化水解速率.图8(a)表明,水解速率随着反应温度的升高而加快.图8(b)为相应催化反应的阿伦尼乌斯曲线(lnK~1/T),通过计算,得到反应活化能(Ea)为35.6 kJ·mol-1,转化频率(TOF)为245.4 mol H2·min-1·(mol Ru)-1.从表2可知,与其他Ru基催化剂相比,催化剂Ru1Mo0.5Sn0.1@MOF-801对 AB 水解制氢具有较低的活化能和较高的转化频率,这主要归功于Ru-Mo-Sn三者之间强的协同作用以及RuMoSn NPs与载体MOF-801间的双功能效应.为了探究催化剂的催化稳定性,测试了催化剂5次循环性能,如图8(c)所示,催化剂经过5次循环后,活性有所降低,这可能与水解过程中溶液的粘度增加或偏硼酸盐浓度增加有关[28].
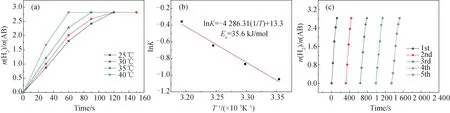
图8 (a) 温度对Ru1Mo0.5Sn0.1@MOF-801催化水解AB的影响曲线; (b) Ru1Mo0.5Sn0.1@MOF-801催化水解AB的阿伦尼乌斯曲线;(c) 催化剂Ru1Mo0.5Sn0.1@MOF-801 5次循环稳定性图

表2 不同钌基催化剂用于AB水解脱氢的催化活性
3 结论
本工作采用水热-液相浸渍法,制备得到Ru1Mo0.5Sn0.1@MOF-801催化剂,将其用于催化氨硼烷水解制氢,催化剂表现出良好的耐久性和稳定性,其反应的活化能为35.6 kJ·mol-1,转化频率值高达245.4 mol H2min-1(mol Ru)-1. Ru1Mo0.5Sn0.1@MOF-801催化剂卓越的催化活性归因于RuMoSn NPs金属间强的协同作用,以及RuMoSn NPs与MOF-801之间的双功能作用.该催化剂制备方法简单,成本低廉,对新型复合催化剂的合成以及氢能源的发展具有重要的参考价值.
猜你喜欢
天然产物研究与开发(2018年7期)2018-08-21 02:04:12
中学化学(2016年2期)2016-05-31 05:27:22
课程教育研究·下(2016年2期)2016-03-25 13:45:48
当代化工研究(2016年5期)2016-03-20 16:21:32
电源技术(2015年11期)2015-08-22 08:50:26
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
低温与特气(2014年4期)2014-03-20 13:36:50
无机化学学报(2014年4期)2014-02-28 17:31:23
应用技术学报(2014年1期)2014-02-28 14:52:11
