
钴基催化剂电催化还原CO2 研究进展
2022-09-02 09:23董仲珍
河北环境工程学院学报 2022年4期
董仲珍,冯 强,郑 鑫
(1. 日照市生态环境保护服务中心,山东 日照 276826;2. 日照市生态环境监测中心, 山东 日照 276826;3. 五莲县环境监测站,山东 日照 262300)
工业化革命以来,人类生产生活对化石燃料的需求与日俱增,不可避免地导致大气中CO2浓度的上升,以及可利用化石燃料储量的下降。 全球二氧化碳排放量已增长到每年约350~400 亿t,随着发展中国家进一步工业化,未来几年可能会继续增加[1]。 CO2被认为是造成温室效应和气候变化的主要气体之一。 根据联合国政府间气候变化专门委员会(IPCC)的预测,到2050 年,全球二氧化碳排放量需要减少50%~85%(与2000 年的水平相比),才能将长期全球平均气温上升限制在2.0~2.4 ℃[2]。 大气CO2浓度升高问题可以通过捕获、封存等技术在一定程度上得到缓解[3]。 然而,CO2的转换、利用或回收方案可能更理想,因为在解决环境问题的同时也满足了对能源的需求。
以CO2为原料的转化过程可以产生多种能源物质如一氧化碳、甲烷、甲醇、乙醇、甲酸和合成气等。 CO2可以通过多种途径进行转化,包括化学、光化学[4]、电化学[5-6]、生物转化[7]。 与其他转化过程相比,电化学还原具有许多优势。 例如,电化学转换可以通过调节施加的电极电位直接控制,反应可以由可再生的风能、太阳能和碳氢燃料生产的电力驱动;此外,电化学系统的占地面积相对较小,在室温运行,化学物质投入量少,并且很容易扩大规模以适应应用[8-9]。 许多具有高催化性能、高选择性、高稳定性和低过电位的贵金属,如Au、Ag、Pt、Pd、Rh 作为催化剂已被用于电催化CO2还原反应(CO2RR)[10]。 然而,贵金属高昂的成本阻碍了它们在CO2RR 中的规模化应用。 钴属于第Ⅷ-B 族元素,作为一种地球丰度较高的过渡金属,其具有高导电性、高催化性能、热稳定性、独特的电子特性和化学稳定性,这使得钴催化剂成为替代贵金属用于CO2RR 的前景材料[11]。 近年来,钴催化剂及其衍生品,包括金属钴、原子钴、钴氧化物、钴酞菁、钴卟啉、钴金属有机配合物等被广泛用于CO2RR。 因此,本研究综述了这些催化剂应用于CO2RR 的研究进展,以期为优良催化剂的制备及碳排放治理提供参考。
1 电催化还原CO2
CO2作为有机物燃烧后的主要产物之一,其电化学还原存在以下几个难点:首先,CO2化学惰性高,分子中的碳原子采用sp 杂化的形式与两个氧原子键合,C=O 双键键长116 pm,碳原子与氧原子形成大π 键。 这一稳定结构赋予CO2分子很高的化学惰性,其在催化位点上的吸附及后续的碳氧键断裂等过程都较为困难。 其次,CO2在水中的溶解度低,在室温、标准大气压条件下,CO2分子在水中的溶解度约为0.034 mol/L,且与碳酸、碳酸氢根、碳酸根等水解产物存在动态平衡,这些产物均难以直接在电催化条件下被还原[12]。 再者,CO2单电子还原为CO2·-的电势较高,在中性水介质中与标准氢电极(SHE)相比达到-1.90 V[13]。 最后,CO2还原涉及多重且复杂的电子转移/质子化耦合过程,因而有许多可能的反应途径,可产生从C1(CO、甲酸盐、甲醇、甲醛和CH4)、C2(乙烯、乙醇、醋酸盐、草酸或草酸盐)到C3(丙醇)的多种化合物,严重限制了反应的选择性[14-15]。
用于CO2电催化还原的设备通常有四个组成部分(图1),包括具有高电导率并允许反应底物和产物快速传质的电解质、减少液体产物氧化的质子膜、涂有高活性和耐用的催化剂的阴极和阳极[16]。 当向电极施加外部电压时,CO2的电催化还原反应发生在阴极的催化剂-电解质界面。
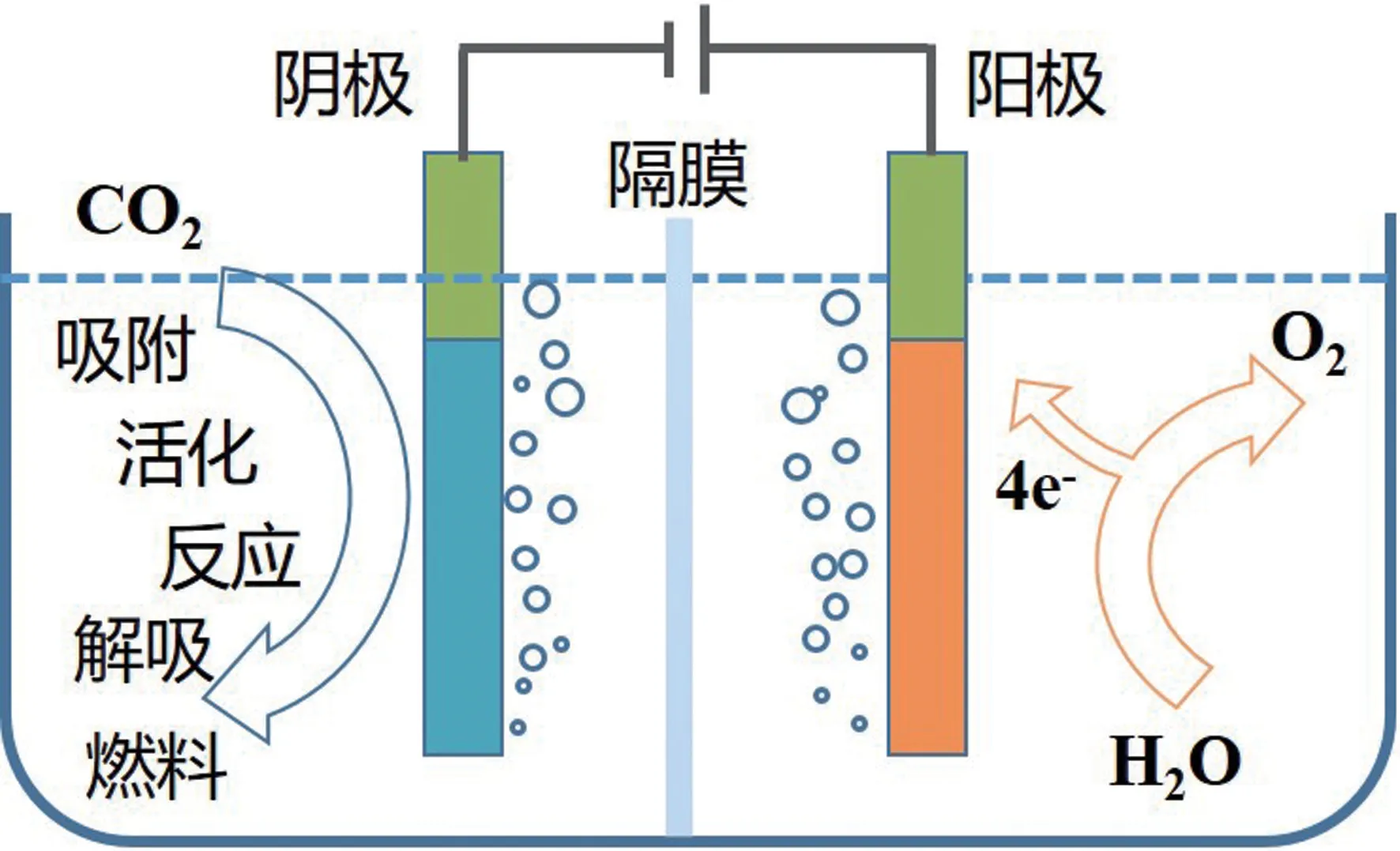
图1 典型的电催化CO2 还原反应池[16]
通常,催化还原过程包括四个主要步骤:首先,CO2在催化剂表面的化学吸附;其次,CO2化学活化为CO2·-;再者,多个电子/质子转移过程导致CO2还原;最后,各种产物从催化剂表面脱附[17]。
2 钴基催化剂电催化还原CO2
2.1 金属钴
金属钴自身具备一定催化性能,当形成纳米颗粒、氮-碳参杂或与其他金属制备合金后,其催化性能将得到提升。 Huang 等[18]合成了分散在单层氮掺杂石墨烯上的钴纳米粒子,在0.1 mol/L NaHCO3电解质中将CO2选择性电催化还原为甲醇,电位为-0.90 V vs.SCE(饱和甘汞电极)时,甲醇的最大法拉第效率(FE)达到71.4%。 Liu[19]合成了Co@ N-C 材料用于CO2RR,暴露在材料表面的Co 主要促进析氢反应(HER)过程产生H2,而由碳-氮骨架包裹的Co 颗粒提供电子并通过Co-NC 骨架将电子传递给CO2分子,促进CO2还原。 酸处理的Co@N-C 在-0.57 V vs.RHE(可逆氢电极)的电位下电催化CO2还原为CO 的FE 仅为60%。 Zhang 等[20]首次通过冷氢气等离子活化法在室温下原位制备了AgCo 合金电催化剂。 电化学测试表明AgCo 对CO2还原产乙醇表现出高催化活性,在电流密度为7.4 mA ·cm-2时乙醇的FE 达到72.3%。 Grote 等[21]发现Cu-Co 薄膜材料在电催化还原CO2方面具有巨大潜力,在催化剂中Co 含量为5%~15%时,CO2倾向于还原形成C2 物质。 张文彪[22]构建了CoxNi1-x合金/碳纳米纤维用于电催化CO2还原。 电流密度达到13.4 mA·cm-2时,在0.5 mol/L NaHCO3电解质中电催化CO2还原为CO 的FE 达到85%。
2.2 单原子钴
单原子催化剂在CO2RR 中具有巨大潜力。Yang[23]通过构建独立的、交联的和高产的碳膜(表示为CoSA/HCNFs)来最大限度地利用单原子钴位点,电流密度为67 mA·cm-2时CO2还原CO的FE 可以达到91%。 He[24]使用第一性原理方法和计算氢电极模型研究了负载在缺陷石墨烯上的单个金属原子电催化CO2还原的性能。 五种过渡金属Ag、Cu、Pd、Pt 和Co 催化剂能普遍能提升C1 产物的生成速率。 Su[25]尝试使用配位数作为控制参数来提高金属的电催化性能。 开发了用配位不饱和3d 金属原子(Co、Ni 或Cu)修饰的共价三嗪骨架(CTF)材料。 Co-CTF 和Ni-CTF 材料有效地将电催化CO2还原为CO 的电位降低到-0. 5 V,CO 的FE 达到90%。
Ren[26]设计了一系列具有明确活性位点电子结构的单Co 原子电催化剂。 实验和密度泛函理论计算结果表明,引入硝基配体具有吸电子能力的单Co 原子催化中心有利于催化CO2还原。 Liu等[27]研究了过渡金属-四氰基醌二甲烷单分子层作为单原子催化剂电催化CO2还原的性能。 结果表明,所研究的十种过渡金属原子对析氢反应具有良好的抑制作用。 Hou[28]通过热解钴金属三联吡啶(Tpy)有机金属配合物合成具有明确位点的单原子Co 催化剂Co-Tpy-C。 Co-Tpy-C 电催化CO2还原为CO 的FE 在电位为-1.0V vs.RHE时超过95%。 Gao 等[29]制造了由纯钴金属以及钴金属和氧化钴的共存形式组成的四原子层催化剂。 电化学测试表明,CO2电还原过程中主要产生甲酸盐(HCOO-)。 原子层的部分氧化进一步增加了它们的内在活性,能够在40 h 测试周期内实现约10 mA·cm-2的稳定电流密度,在仅0. 24 V 的过电位下具有约90%的甲酸盐选择性。
2.3 钴氧化物
氧化物Co3O4似乎是电催化CO2还原最具竞争力的候选材料之一。 Co3O4具有环境友好、成本低、储量丰富等特点;此外,其结构为非正态尖晶石结构,四面体间隙中含有CO2+离子,而八面体间隙中包含Co3+离子,其独特的晶体结构使其具有较高的环境稳定性[30]。
Yadav 等[5]探讨了Co3O4作为阳极电催化CO2还原的性能。 在0.5 mol/L KHCO3溶液中,1.5~3.5 V 电压下进行电催化测试时,CO2主要还原为乙醇,在2 V 电压下反应5 min,乙醇的最大FE 为96.15%。 此外,在石墨板上沉积合成的Zn和Co3O4电极,分别用作阴极和阳极用于电催化CO2还原。 液相色谱检测表明HCOOH 是在该条件下形成的唯一还原产物。 在电压为1.5 V 时的KHCO3电解质溶液中反应10 min 后,HCOOH 的最大FE 为78.54%[31]。 Aljabour[32]进行了半导体Co3O4纳米纤维电催化CO2还原的探索,CO2转化为CO 的FE 为65%。 Zhang 等[33]通过在Co3O4-CeO2/低石墨碳(Co3O4-CeO2/LGC)上制造氧空位来降低电催化CO2还原生成甲酸盐的过电位。实验结果表明,在Co3O4-CeO2/LGC 电极上CO2转化为甲酸盐的过电位仅为0.31 V vs.RHE,在电位为0.75 V vs.RHE 时,甲酸盐的FE 为76.4%,并能稳定运行45 h。 Gao 等[34]探究了氧空位在钴单晶胞层电催化CO2还原中的作用。 在40 h 测试周期内,富含空位的氧化钴单晶胞层具有2.7 mA·cm-2的电流密度,甲酸盐的FE 为85%。
2.4 钴酞菁
钴酞菁(CoPc)是钴的大环配合物,其分子由一个酞菁(Pc)环和中心的Co 金属离子组成[35]。早在1974 年,Meshitsuka 等[36]就探究了吸附在石墨上的CoPc 电催化CO2还原的性能。 为了实现CoPc 的高稳定性与催化选择性,一系列措施被用来提升其催化性能,包括固定在碳基材料如活性炭、碳纳米管、石墨、石墨烯;基团取代,碳-氮参杂,聚合钴酞菁等。 Zhang 等[37]制备的钴酞菁/碳纳米管(CoPc/CNT)复合催化剂,在电流密度为15.0 mA·cm-2,电位为0.52 V 时,电催化CO2还原为CO 的FE 效率为98%,转换效率(TOF)为4. 1 s-1。 Chen 等[38]将聚合CoPc 催化剂负载在CNT 上作为工作电极,CoPPc-CNT 的起始电位为-0.35 V vs.RHE 时,CO 的FE 达到96%。 马静静[39]制备了碳-氮参杂钴酞菁(CoPc/N-C)用于电催化CO2还原,在0.1 mol/L KHCO3电解液中进行电化学测试,电位为-1 V vs.RHE 时,CO 的FE>85%,其优异的电催化性能归因于复合材料中CoPc 与N-C 载体之间的协同作用。
Gu 等[40]使用石墨二炔/石墨烯(GDY/G)异质结构作为二维导电支架来锚定CoPc。 在H 电池中进行电化学测试,在电位为-1.0 V vs.RHE,电流密度为12 mA·cm-2时,CO 的FE 为96%,TOF 达到37 s-1。 类似地,石墨烯中引入丰富的本征缺陷可以促进石墨烯基底和CoPc 之间的π电子转移,产生更多的电催化活性Co 位点并导致CO2+/Co+还原电位正移,从而增强了CO2化学吸附[41]。 于学政[42]将单层分散的CoPc 耦合在缺陷石墨烯上制得CoPc/DG 复合材料,CO 的FE 在电位为-1.08 V vs.RHE 时达到94%,而H2的FE 仅为5%。 经过11 000 s 的连续催化反应,CO 的FE仍保持在93%。
2.5 钴卟啉
钴卟啉作为另一种较为高效的CO2RR 催化剂,通过外围功能化或异质化同样可提高其催化性能。 Pander[43]使用简便的电聚合工艺制备了用钴卟啉薄膜修饰的电极。 该电极在水溶液中可将CO2电催化还原为CO,在过电位为450 mV 时,FE 达到(84±2)%。 四苯基钴卟啉(CoTPP)可被用作均相和非均相条件下的催化剂。 将CoTPP直接固定在碳纳米管上后电催化能力显著增强,在水介质中,电催化CO2还原为CO 的FE 大于90%[44]。 Zhang 等[45]将两种带相反电荷的钴卟啉固定在碳上并在350 ℃下的温和热解,使钴卟啉失去一些外围基团并紧密吸附在碳上。 在430 mV 的低过电位下将CO2电催化还原为CO,FE 和电流密度分别为88%和8 mA·cm-2。 Zhu等[46]在石墨烯载体上引入氮原子后,钴卟啉/石墨烯复合材料的催化活性提高了两倍。 在低容量三电极电池中测试了电催化CO2还原的电化学性能,在电位为-0.4 V vs.RHE 时,CO 的FE 达到80%。
Dou 等[47]通过在钴卟啉配体上添加更多的芳族取代基来扩大π 共轭,改变的π 电子体系赋予了所制备的钴卟啉更强的催化性能。 在电位为-0.6 V vs.RHE 时,CO 的FE 高 达95%,TOF 为2.1 s-1。 此外,TiO2也被用作固定四苯基钴卟啉(CoTPP)的载体,电化学测试结果发现TiO2的晶相明显影响CO2的电催化还原。 锐钛矿相比金红石相表现出更高的活性和选择性,因为增强的电导率反过来使载体和CoTPP 之间的电子转移更快[48]。
2.6 钴基金属有机框架/共价有机框架
金属有机框架(MOF) 和共价有机框架(COF)的网状化学可调控二氧化碳转化为增值产品[49]。 然而,这种可调特性被较差的导电性所阻碍。 通过在无机组分中适当使用钴、铜等金属取代可克服上述问题。 Meng[50]通过铜和金属酞菁配位制备的导电二维MOF 具有高电导率,合成的CoPc-Cu-NH 和CoPc-Cu-O 电催化CO2还原为CO 的TOF 分别为1.15 s-1和0.63 s-1。 Lin[51]以钴卟啉作为结构单元,通过亚胺键键合形成COF用于电催化CO2还原为CO。 随着链长度的增加,CO 的TOF 高达2.6 s-1,比正常钴络合物高约26 倍。 Han 等[52]制备了二维聚酰亚胺连接的酞菁COF(表示为CoPc-PI-COF-1 和CoPc-PICOF-2)。 在0.5 mol/L 的KHCO3电解质中,在-0.80 V vs.RHE 的电位下,CoPc-PI-COF-1 和CoPc-PI-COF-2 电催化CO2还原为CO 的FE 分别为97%和96%。 然而,在电位为-0.90 V 时,CoPc-PI-COF-1 表现出更大的电流密度(-21.2 mA·cm-2),和TOF(2.2 s-1)。
Zhang 等[53]将钴卟啉精确锚定在二维MOF纳米片(Zr-BTB)上获得超薄二维MOF 纳米片(TCPP(Co)/Zr-BTB)用于电催化CO2还原。 与分子钴卟啉相比,合成的催化剂由于活性位点利用率高,在电位为-0.92 V vs.RHE 时电催化CO2还原为CO 的TOF 高达1.32 s-1。 通过对氨基甲基苯甲酸(PABA)、对磺基苯甲酸钾(PSBA)和对氨基苯甲酸(PSABA)改性后的二维MOF 纳米片TCPP(Co)/Zr-BTB-PABA、TCPP(Co)/Zr-BTBPSBA、TCPP(Co)/ZrBTB-PSABA 中TCPP(Co)/Zr-BTB-PSABA 表现出最佳电催化性能,在电位为-0.769 V vs.RHE 时,电催化CO2还原为CO 的FE 和TOF 分 别 为85.1% 和1.48 s-1。 Matheu等[54]制备了一种含有钴酞菁的MOF 用于电催化CO2还原,在电位为-0.52 V vs.RHE 时,电催化CO2还原为CO 的FE,电流密度和TOF 分别为80%,-16.5 mA·cm-2,和0.20 s-1。 Zhang 等[55]合成了一种新型混合金属MOF(Ag4CO2(PYZ)PDC4)用于电催化CO2还原,催化剂在0.1 mol/L KHCO3电解质中电催化CO2还原为CO 的FE 为55.6%。
3 结论与展望
钴形成的一系列催化剂降低了CO2电催化还原过程的反应能垒,从而实现CO2向C1、C2、C3、C4 产物转化。 固定、包埋、基团取代等手段可以提高钴氧化物、钴卟啉、钴酞菁、钴基金属有机框架/共价有机框架的电催化性能和稳定性。 目前,钴基催化剂已实现了电催化CO2转化成CO 或甲酸等C1 产物,但较难实现CO2直接向C2、C3 产物转化。 尽管目前的研究取得了较大进展,但仍然存在以下挑战:
(1)电化学CO2还原涉及多个电子/质子耦合过程,并且在固定的过电位上可能存在不同的反应途径。 提高催化剂的选择性仍然是一个挑战。
(2)电化学CO2还原产物包括气态和液态,产物的快速解吸会破坏微观结构并阻碍电解液扩散。 此外,催化剂的活性位点也可能被电解液中的反应中间体、副产物和杂质堵塞。 迄今为止报道的催化剂的稳定性尚未达到1 000 h。
(3)电催化CO2还原过程常伴随析氢反应发生,如何抑制副产物的生成和目标产物的纯化是难点。
(4)目前的研究都是基于实验室控制手段,自然大气条件组分复杂,从大气中如何分离CO2并高效吸附于催化剂用于还原实验的数据缺乏。
(5)近年来,共价有机框架(COF)和共价有机框架(MOF)的合成方法和应用取得了快速发展。 然而,高度稳定的功能性COF 的合成仍然是一个巨大的挑战
未来,需要进一步探索的方向有:
(1)加强催化剂与CO2分子的结合是非常必要的。 没有CO2分子的结合位点,CO2不能集中在催化剂周围,催化中心难以接触、活化和转化CO2分子。 因此,非常需要在催化剂中引入CO2亲和性官能团。 特别是,如果空间位阻可以忽略不计,则预计此类官能团将位于金属中心附近。据报道,具有-OH 和-NH2基团的配体有利于CO2捕获。 其次,金属中心周围配体上的配位原子的路易斯碱度也会影响催化剂的催化性能。 原则上,配位原子的弱路易斯碱度容易导致金属中心被还原,这将降低电催化CO2还原的过电位。
(2)金属配合物为开发CO2还原催化剂提供了完善的研究平台。 但仍然需要使用有机金属配合物来增加材料的共轭性,以克服高于八电子体系催化剂的电导率问题。
(3)通过实际应用了解催化剂的活性位点、还原途径和中间体类型至关重要。 空气中的CO2与其他气体混合存在,必须测试存在其他气体组分时催化剂电催化CO2还原的性能以揭示催化剂实际应用的活性和稳定性。
(4)钴基催化剂使用带有饱和CO2的液体电解质进行还原反应,这会导致电催化CO2还原过程中电流密度过低,可以通过引入气体扩散电池来克服液体电解质的溶解度问题并实现高电流密度(>100 mA·cm-2)。
猜你喜欢
油气田地面工程(2022年8期)2022-10-02
蓄电池(2022年1期)2022-02-25
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
表面工程与再制造(2019年6期)2019-08-24
广东教育·高中(2018年12期)2018-02-13
分析化学(2018年12期)2018-01-22
科技创新与应用(2017年11期)2017-04-27
分析化学(2017年1期)2017-02-06
中小企业管理与科技·下旬刊(2016年12期)2017-01-17
