
伴有皮质下梗死和白质脑病的常染色体显性遗传性脑动脉病1例
2022-08-29 04:27朱源义
武警医学 2022年8期
关 鉴,李 可,强 薇,张 巍,朱源义
伴有皮质下梗死和白质脑病的常染色体显性遗传性脑动脉病(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy,CADASIL),是一种较为少见的遗传性小动脉病,由位于19号染色体上的NOTCH3基因中的半胱氨酸错义突变所引起。成年发病,多表现为偏头痛、反复卒中及进行性认知功能障碍,并伴有精神症状,最终导致严重的残疾和痴呆。由于该病缺乏特异性,易误诊、漏诊,且预后较差。本文通过对1例CADASIL患者的临床表现及影像学特点进行分析,并复习相关文献,以期为临床早期准确诊断 CADASIL 提供参考。
1 病例报告
患者,男,48岁。3年前,家属发现其注意力减退,反应迟钝,动作完成缓慢,少言,淡漠,易怒。20 d前,患者突发言语不清,吐词缓慢、 含糊,感左侧肢体乏力,第2日达高峰,乏力最为明显,行走时向左侧倾斜。家族史:父亲有“偏侧无力”史,平日颈部低垂,动作迟缓,反应迟钝,60岁时因“重症肌无力”去世;叔叔因“脑出血”于60岁去世;姑姑患“痴呆”,大小便失禁,长期卧床;姐姐患有“脑梗死”,治疗后可自行行走,日常生活基本自理。专科查体:神志清楚,吐词含糊不清,言语缓慢,反应稍显迟钝,理解力、判断力正常,定向力、执行力、记忆力、计算力下降。颅神经(-),四肢无局限体征。同型半胱氨酸(32.4 μmol/L)中度升高。
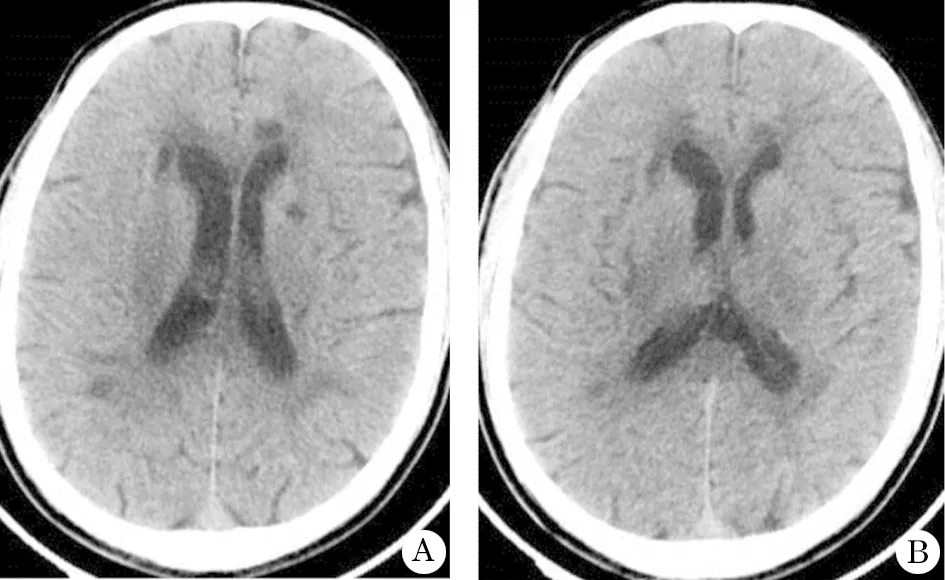
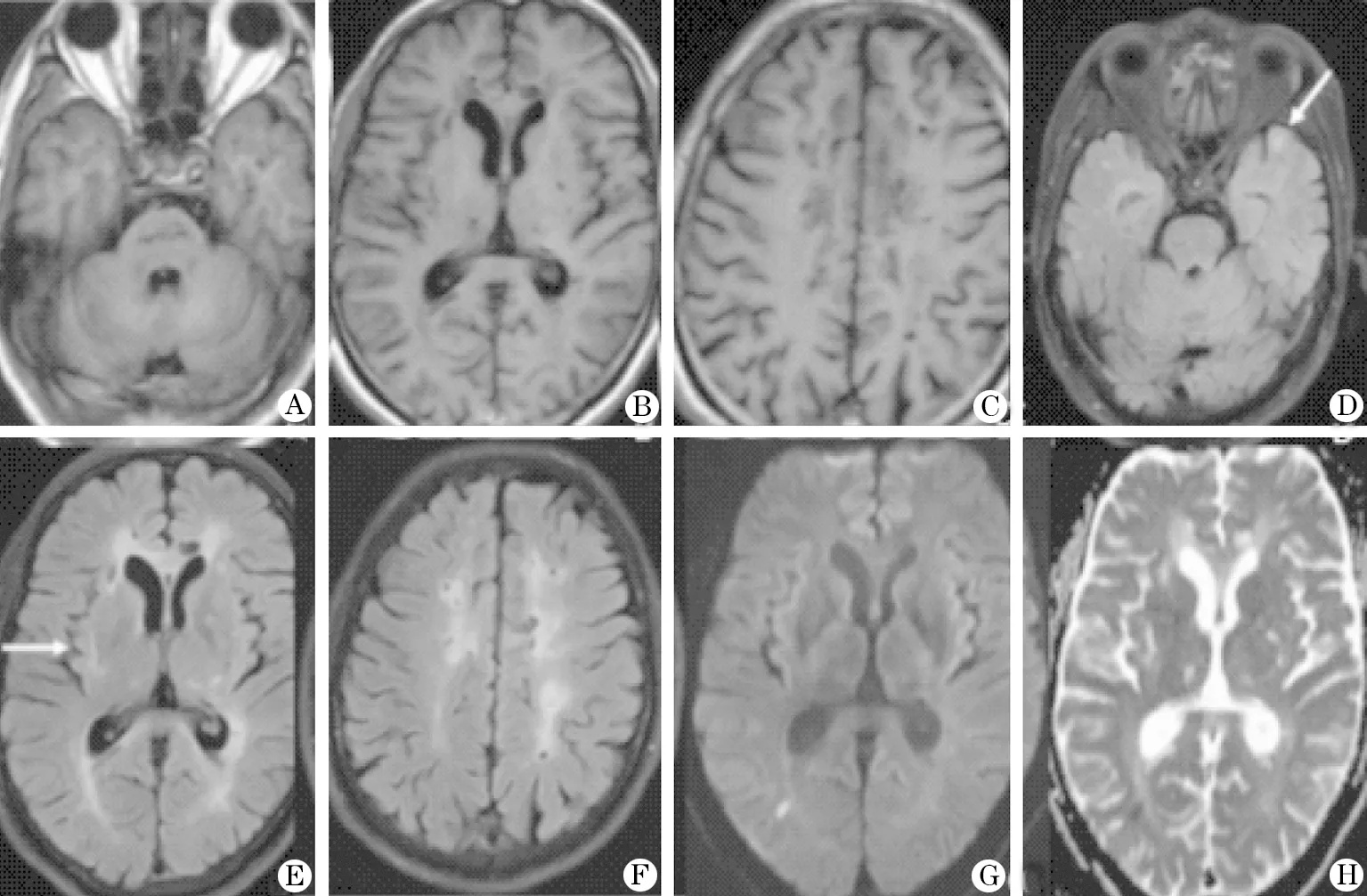
头部CT(图1)提示:双侧基底节区、侧脑室旁及放射冠区多发小结节状低密度影,考虑为脑腔梗灶。头部 MRI(图 2)提示:脑桥、双侧基底节、丘脑、侧脑室周围、半卵圆中心及胼胝体区多发长T长T信号影,FLAIR序列上呈低信号,周围见高信号环绕,考虑为多发陈旧性腔隙性脑梗死;此外FLAIR序列上双侧颞极(颞前叶)及外囊区见对称性斑点状高信号影;右侧枕叶见少许点状弥散受限灶,考虑为急性皮层下小梗死;双侧侧脑室周围脑白质脱髓鞘改变;轻度脑萎缩。
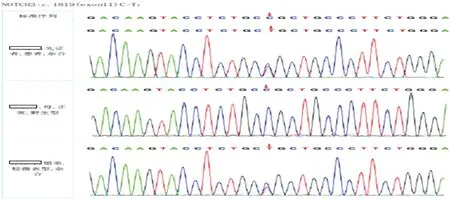
基因检测:在签署知情同意书后,采集先证者、先证者之母、先证者之姐弟的血液标本进行全外显子测序。测序结果(图3)提示:患者及其姐弟均存在Notch3基因第11号外显子杂合突变:c.1819C >T,导致氨基酸改变:p.Arg607Cys,其母正常。
2 讨 论
2.1 临床表现及病理学特征 CADASIL临床外显率与年龄有关,发病平均年龄为45岁,无性别差异,患者多无高血压、糖尿病病史,常有家族遗传史。主要表现为偏头痛、皮层下缺血事件、认知功能障碍和情感障碍,且大致随病程进展而演变。偏头痛通常为CADASIL首发症状,且多为有先兆性偏头痛。脑梗死或短暂性脑缺血发作是最常见的临床症状,表现为纯运动性卒中、共济失调性轻偏瘫、
CADASIL的病理学表现可概括为系统性小动脉病变、大脑深部多发腔隙性脑梗死和广泛白质脱髓鞘改变。电镜下可见受累血管中层内颗粒状电致密度嗜锇物质(granular osmiophilic material,GOM)沉积,围绕在平滑肌细胞周围;在脑、脾、肝、肾、肌肉和皮肤小血管及颈动脉、主动脉壁均可出现类似改变。上述部位活检发现GOM沉积是诊断CADASIL的特异性标准。
CADASIL是较少见的单基因显性遗传性脑动脉病,是由位于19号染色体短臂上的Notch3基因突变所致,目前已报道了300多种形式的Notch3基因突变类型。Notch3有33个外显子,而发现的CADASIL突变基因都集中在2~24号外显子中。不同地区和种族的NOTCH3基因突变位点存在较大差异,我国最常见的突变位点是第3、4号外显子,其次位于第11、12、13、14、18外显子。本病例基因突变位于第11号外显子,突变位点:c.1819C>T p.(Arg607Cys),符合CADASIL的基因突变特征。
2.2 影像学特征 CT检查缺乏特异性,可表现为脑内多发散在的边界清晰的腔隙性脑梗死灶。MRI对CADASIL的诊断具有较高的敏感性和特异性,主要征象包括白质高信号、腔隙性脑梗死和脑微出血灶。其中白质高信号表现为TWI及FLAIR序列上对称分布于双侧侧脑室周围和深部脑白质的异常高信号,其最初累及半卵圆中心,随病程进展逐渐扩展至双侧颞极、外囊和胼胝体。颞极白质高信号(O’Sulliva征)和外囊横轴位高信号(“人”字征)是诊断CADASIL的特征性影像学标志,其敏感性分别为89%和86%,特异性分别为93%和45%。CADASIL患者脑内可见多发腔隙性脑梗死灶,分布于半卵圆中心、基底节、丘脑及脑干,急性期缺血性脑梗死于DWI上呈弥散受限改变,非急性期病灶于MRI各序列中均类似于脑脊液信号。有研究发现,腔隙性梗死灶的数量和面积与特定的执行功能以及总体认知功能呈负相关,提示腔隙性梗死灶还可作为预测CADASIL认知功能损害的重要因素之一。脑微出血灶好发于皮层-皮层下区域、白质、丘脑和脑干,在T2WI梯度回波序列和磁敏感加权序列中呈均匀一致的点状低信号影,其发生率为31%~69%。
2.3 诊断 CADASIL发病率较低,临床表现无特异性,影像学特点未被重视,确诊检查如皮肤活检或基因检测等较复杂,因此,临床中易被误诊为普通偏头痛、脑梗死、脱髓鞘脑病、痴呆或精神异常等。目前国内CADASIL诊断标准包括:(1)发病情况:中年起病,常染色体显性遗传,多无高血压、糖尿病、高胆固醇等危险因素;(2)临床表现: 脑缺血性小卒中发作、认知障碍或情感障碍等表现中的1项或多项;(3)颅脑MRI:大脑白质对称性高信号病灶,颞极和外囊受累明显,伴有腔隙性脑梗死灶;(4)病理检查:血管平滑肌细胞表面GOM或Notch3蛋白免疫组化染色呈阳性;(5)基因筛查检出Notch3基因突变。满足前3条和第4或第5条为明确诊断;满足前3条为可疑诊断;满足前2条为可能诊断。
2.4 鉴别诊断 CARASIL是HTRA1基因突变所致的遗传性脑小血管病;青少年起病,患者多有明显的秃顶,腰背部疼痛,脊柱关节变形僵硬,20~30岁发病,痉挛性步态异常为最常见始发临床表现,随后缓慢进展并出现神经系统其他症状;CARASIL的脑部MRI表现与CADASIL极为相似,但前者极少有颞叶前部和外囊受累,白质改变从疾病早期就均匀性融合发生;病理学检查未发现有嗜锇颗粒物或淀粉样蛋白沉积。皮层下动脉硬化脑病常在中老年人分散发病,阶梯性发展痴呆以及脑卒中反复发作、长期高血压病史。病理学检查可发现小动脉硬化导致的内膜肥厚,血管平滑肌细胞表面无GOM。MRI表现为侧脑室周围白质弥漫性损害,也可发现基底节、丘脑、脑干梗死改变,一般没有双侧颞极的白质损害。
综上所述,如果患者发病年龄小于50岁,缺乏脑卒中的常见危险因素,有反复发生的皮层下缺血性卒中、严重的情感障碍、伴先兆的偏头痛、痴呆等表现,MRI上可见特征性的双侧颞极和外囊高信号,需考虑遗传性脑小血管病CADASIL的可能性,再追查家族史,结合皮肤活检和基因检测,予以确诊。
猜你喜欢
保健与生活(2019年15期)2019-09-12
中国社区医师(2018年6期)2018-11-20
中学生理科应试(2017年6期)2017-09-27
中学生理科应试(2017年2期)2017-04-01
中外医疗(2016年33期)2017-03-02
东西南北(2016年17期)2016-10-14
中国医药科学(2014年24期)2015-03-16
心脑血管病防治(2014年6期)2015-01-20
