
原子吸附对二维GeSe光电性质的影响
2022-08-24 12:52:50张国英管永翔
沈阳师范大学学报(自然科学版) 2022年3期
张国英, 于 乐, 管永翔
(沈阳师范大学 物理科学与技术学院, 沈阳 110034)
0 引 言
近年来,人们对二维材料的研究取得了很大进展。2004年,英国物理学家Novoselov等[1]成功地在实验中从石墨上分离出了石墨烯,并证实它可以单独存在。石墨烯因其超高的物理性能,在过去的十几年中成为最受关注的材料[2]。石墨烯在纳米技术和纳米器件领域的成功应用开启了对层状低维材料的广泛探索和研究。但随着人们对二维材料的深入研究,石墨烯的缺点也渐渐显露出来,它的零能隙成为其在光学器件应用上的短板。为了找出更适合应用于光电器件的材料,人们开始研究单层单硫族化合物[3-5]。单层单硫族化合物二维材料[6-9]因其优异的性能和较强的抗氧化能力而受到人们的关注。锗或锡的单硫族化合物具有锗型斜方晶结构(空间群为Pnma),与黑磷烯结构类似[10]。二维硒化锗(GeSe)材料已被证实在光电领域很有应用潜力,因此,对GeSe二维化合物进行系统研究非常必要[11]。到目前为止,基于GeSe纳米片的光电行为的实验研究已有一些报道。有实验表明,具有良好光学带隙的少层GeSe薄片可以有效地收集太阳光,用于基于光电化学电池的光电流产生。原子厚度的GeSe薄片是一种新的二维半导体,在光电子和光子学领域有潜在应用。这些有趣的特性使GeSe单层膜成为一种有前途的用于纳米力学、热电学和光电子学的二维材料。理论研究发现采用HSE06[12-13]杂化泛函并进行vdW校正可得到GeSe的比较精确的电子结构信息。二维结构导致能带结构上出现非常小的色散,态密度中出现尖锐的峰,这些特点与强光吸收有关,有利于光伏应用[14-16]。但在含缺陷、掺杂或缺陷、掺杂与应变结合等复杂情况下对GeSe光电性能的调控研究还较少。为了进一步掌握GeSe光电性能的影响规律及机理,本文对GeSe二维材料的光电学性质的影响规律及机理进行了系统研究,希望为GeSe二维材料在光电领域的应用提供理论依据。
1 计算模型及理论方法
GeSe在理论上被认为是唯一具有直接带隙的材料,且该材料的光谱范围预测几乎覆盖了整个太阳光光谱,这使得它在量子光学、光电探测、光伏、电学等领域有巨大的应用潜力[17]。GeSe晶体具有层状结构,属于正交晶系,空间群为Pnma(No.62),实验上获得的晶格参数为:a=3.878 Å,b=4.550 Å,c=11.142 Å,键角α=β=γ=90°。对单层GeSe选取(1×1×1),(2×2×1),(3×3×1)超原胞进行了收敛性测试计算。为防止GeSe层间的相互干扰,在C轴方向创建15 Å的真空层。图1给出了(2×2×1)GeSe单层俯视图、正视图及侧视图。
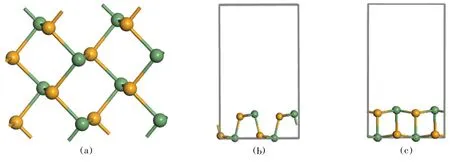
图1 (2×2×1)GeSe单层(a)俯视图;(b)正视图;(c)侧视图Fig.1 (a)Top view; (b) front view; (c)side view of (2×2×1)GeSe monolayer
为了研究二维GeSe材料的光电性质,应用具有超软赝势的CASTEP软件包实现密度泛函理论(density functional theory,DFT)计算,包括几何结构弛豫和电子结构及光学性质的计算。本文首先用2种策略分别优化了GeSe(Pnma)晶体的结构,得到带隙和晶格参数来确定交换关联函数,比较广义梯度近似(generalized gradient approximation,GGA)和局域密度近似(local density approximation,LDA),结果发现GGA的计算结果与实验值更接近;其次,对GGA的不同形式的交换关联函数进行比较,如对PW91,PBE和PBESOL等进行了测试和比较,发现PW91计算的晶格参数(a=4.41 Å,b=3.84 Å和c=10.82 Å)与之前的理论和实验结果更接近[1]。因此,以下主要利用这个势函数来计算GeSe单层的光电性质。
确定了交换关联函数GGA-PW91后,要确定优化GeSe单层的计算参数,包括平面波展开的截断能和布里渊区K网格的设置。选(1×1×1)GeSe单层,取不同截断能和K值计算带隙和晶格参数a,b,c,计算结果列于表1。GeSe单层材料能隙的实验值约为Eg=1.0~1.3 eV,晶格参数的实验值是a=4.16 Å,b=3.96 Å,c=19.93 Å[18]。从表1可以看出,不考虑自旋,截断能取600 eV,k网格设置为4×4×1, 5×5×1时,计算的带隙值明显大于实验值。故k网格设置为3×3×1。这样设置,使得截断能增大,带隙值增大,晶格参数a稍有降低,b,c有所增加。与实验结果相比较发现,截断能取310和350 eV时最好。本文还研究了自旋对带隙及晶格参数的影响,发现当k网格设置为3×3×1, 截断能设定为350 eV时,考虑和不考虑自旋对结果影响不大。因此,后续的计算不考虑自旋,k网格设置为3×3×1,截断能取350 eV。关于超原胞的选取,取(1×1×1),(2×2×1),(3×3×1)GeSe超原胞,计算带隙和晶格参数,结果发现,(3×3×1)GeSe单层的带隙只比(2×2×1)单层大0.016 eV(表1),而密度泛函理论能量精度就是0.1 eV,所以为了节省计算资源,后续光电性质的计算超原胞均选(2×2×1)单层,原胞有8个Ge和8个Se原子。在自洽场计算中,能量的收敛精度为5.0×10-5eV·atom-1, 每个原子上的受力不大于0.1 eV·nm-1, 内应力收敛精度为0.2 GPa,原子最大位移收敛标准为0.005 Å。

表1 不考虑自旋极化、取不同截断能和设置不同k点时计算的(1×1×1),(2×2×1),(3×3×1)GeSe单层的能隙和晶格参数
为了分析单层 GeSe吸附原子前后的光学性质, 可以利用计算占据态和非占据态波函数的矩阵元素得到复介电函数虚部ε2(ω),介电函数实部ε1(ω)则可以利用Kramers-Kronig色散关系求出[19]。所有其他的光学性质便可以由ε1(ω)和ε2(ω)推导出,如反射系数、吸收系数等。
复介电函数表达式[20]为
其中:m为自由电子质量;C,V分别表示导带和价带;BZ代表第一布里渊区;k是消光系数;|e|MCV(K)|为动量跃迁矩阵元,K是倒格矢;虚部ε2(ω)是描述占据电子态和未占据电子态之间的真实跃迁;实部ε1(ω)反映了激发态的电子释放能量向低能级的跃迁。其他光学常数,包括吸收系数I(ω)、折射率n(ω)、消光系数k(ω)、反射率R(ω)等均可由介电函数的实部ε1(ω)和虚部ε2(ω)推导得到,具体关系式[21]如下所示:
上述关系式是分析能带结构和光学性质的理论依据,尤其介电函数虚部ε2(ω)是能带结构的重要体现形式[22]。
2 结果分析与讨论
2.1 吸附能结果讨论
通常情况下,吸附的存在会影响材料的稳定性,吸附能可以反映吸附原子与吸附剂的结合强度。吸附能Ef由下式给出:
Ef=(EGeSe+Eatom)-Eatom/GeSe
(8)
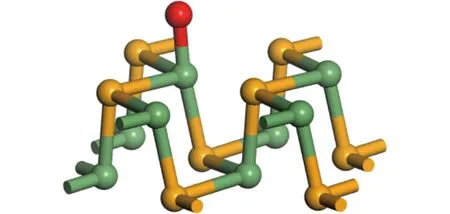
图2 GeSe单层上吸附原子的位置Fig.2 The positions of absorped atom on GeSe single layer
其中:EGeSe代表未吸附原子时单层GeSe的能量;Eatom/GeSe表示吸附原子后体系的总能量;Eatom代表吸附原子的能量。根据吸附能公式可知,要形成稳定的吸附,吸附原子后总能量要降低,所以Ef为正值的吸附能自发进行。根据上式计算出(2×2×1) GeSe单层在顶位Ge原子正上方(吸附位置如图2所示)吸附O,Al或B原子的吸附能,见表2。从表2可以看出,当GeSe单层吸附O,Al或 B原子时,吸附能都为正,即都能形成稳定的吸附。当吸附氧原子时吸附能最大,可见O原子与GeSe单层的结合最稳定。

表2 GeSe吸附O原子、Al原子或B原子的吸附能Table 2 Adsorption energy of GeSe adsorbing O atom, Al atom or B atom
2.2 能带结构与态密度
本研究首先计算了GeSe单层和吸附Al,B,O原子的GeSe单层(图2)的能带结构(图3)和态密度(图4)。从图3(a)可以看出,未吸附原子的GeSe单层带隙宽Eg约为1.213 eV(Eg=E最低非占据态-E最高占据态), 导带底和价带顶都出现在能带图中从G到F的路径上,说明GeSe是直接带隙半导体,这与Hu等[23]的研究结果一致。GeSe单层带隙的实验值为1.1~1.3 eV,可见本文的结果与实验值比较符合。吸附O原子后,O原子在纯净GeSe单层的带隙中接近导带底处产生一个杂质能带,这个杂质能带成为了最低的非占据态,于是GeSe单层的带隙变窄,为1.092 eV(图3(b))。B原子被吸附后,在纯净GeSe单层的带隙深处产生一个杂质能带,此时费米能级位于这个能级以上的位置,于是这个杂质能带成为了最高的占据态,进而GeSe单层的带隙变得非常窄,为0.283 eV(图3(c))。吸附Al原子产生的杂质带跨越了费米能级,使得占据态和非占据态连续分布,于是吸附Al的GeSe单层带隙变为0 eV(图3(d))。从整体来看,O原子的吸附对带隙的影响较小,B原子以及Al原子的吸附可以大大降低GeSe单层的带隙甚至使带隙变为0。此外,从图3还可以看出,纯净的和掺杂O的GeSe单层的费米能级位于价带顶,是P型半导体;掺杂B的GeSe单层费米能级处于导带底部,是n型半导体;掺杂Al的GeSe单层费米能级跨越杂质带使GeSe单层变为了导体。带隙的变化与GeSe光学性质的变化有十分密切的关系。众所周知,半导体吸收限光子的能量(最小能量)hν=Eg, 纯净的GeSe单层带隙为1.213 eV,对应吸收光的频率为2.93×1014Hz, 波长为1 022.3 nm,处于红外区,说明整个可见光谱都在吸收范围内。吸附原子后,带隙宽度减小,其吸收限向低频率方向偏移,增大了GeSe单层的光吸收范围。
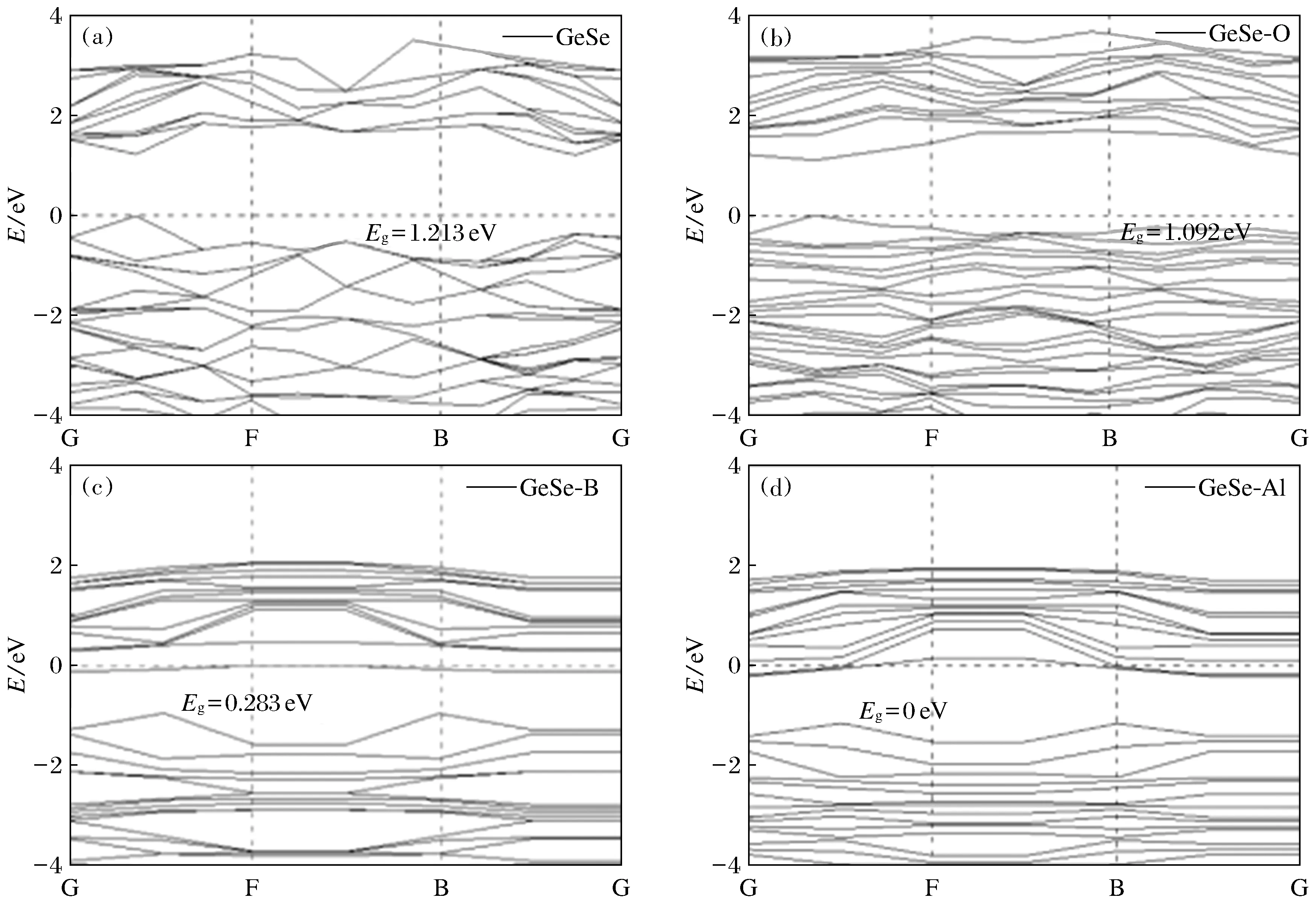
(a) 本征GeSe; (b) 吸附O原子; (c) 吸附B原子; (c) 吸附Al原子图3 GeSe单层的能带结构Fig.3 Energy band structures of GeSe monolayers
图4为本征、吸附O,B,Al原子的GeSe单层的总态密度图和吸附Al,B,O 原子及其近邻的Ge,Se的分波态密度, 费米能级位置取为能量零点。由图4(a)可见,纯净的和吸附O的GeSe单层的带隙值与能带结构图给出的带隙值接近,费米能级位于价带顶,是P型半导体。结合图4(d)可知吸附O产生的能态主要集中在价带,并未在带隙或能带底或能带顶产生杂质能带。吸附的B和Al在带隙中产生杂质带,由于在态密度绘图时有高斯展宽,所以杂质带与导带连到了一起,可见带隙很小。由图4(b)可以看出,导带底的杂质带主要是Al的p电子贡献的,Al的s电子和Ge的p电子也有一定的贡献。由图4(c)可以看出,导带底的杂质带主要是B的s电子贡献的。吸附的B和Al的GeSe单层的费米能级在导带底,由此可推测出吸附B和Al原子的GeSe单层是n型半导体或导体(由能带结构图可知吸附Al原子的GeSe单层是导体)。吸附原子对带隙的调制及半导体类型的改变为GeSe单层在电子器件领域的应用提供了思路。
2.3 光学性质
图5为GeSe单层吸附不同原子时折射率和消光系数随能量的变化曲线。由图5(a)可以看出,GeSe单层的静态折射率(R(0))为2.40,吸附Al,B,O原子后GeSe单层静态折射率分别为5.80,2.40,2.25。吸附Al原子的GeSe单层在能量小于1.0 eV时有较大的值,大于1.0 eV的折射率变化趋势与纯净的GeSe单层类似。吸附B和O原子对GeSe单层折射率变化趋势影响不大,折射率首先在低能区随着能量增大到峰值2.55~3.0(能量在约2.0 eV处),然后在7.5 eV附近减小至最低,接近于零,然后再增大,能量大于8 eV时折射率趋于平缓, 折射率约为0.8~0.9。由图5(b)可以看出,吸附Al原子的GeSe单层的消光系数与纯净和吸附B,O原子的GeSe单层明显的区别是在能量约为0.50 eV时出现第一个峰值,约为1.5。在高于2.0 eV能区,各种GeSe单层消光系数变换趋势相同。最大峰值出现在能量为3~5 eV区域,其中吸附Al原子的GeSe单层的消光系数最大峰值为2.15,吸附O原子的GeSe单层的消光系数峰值最低为1.78。
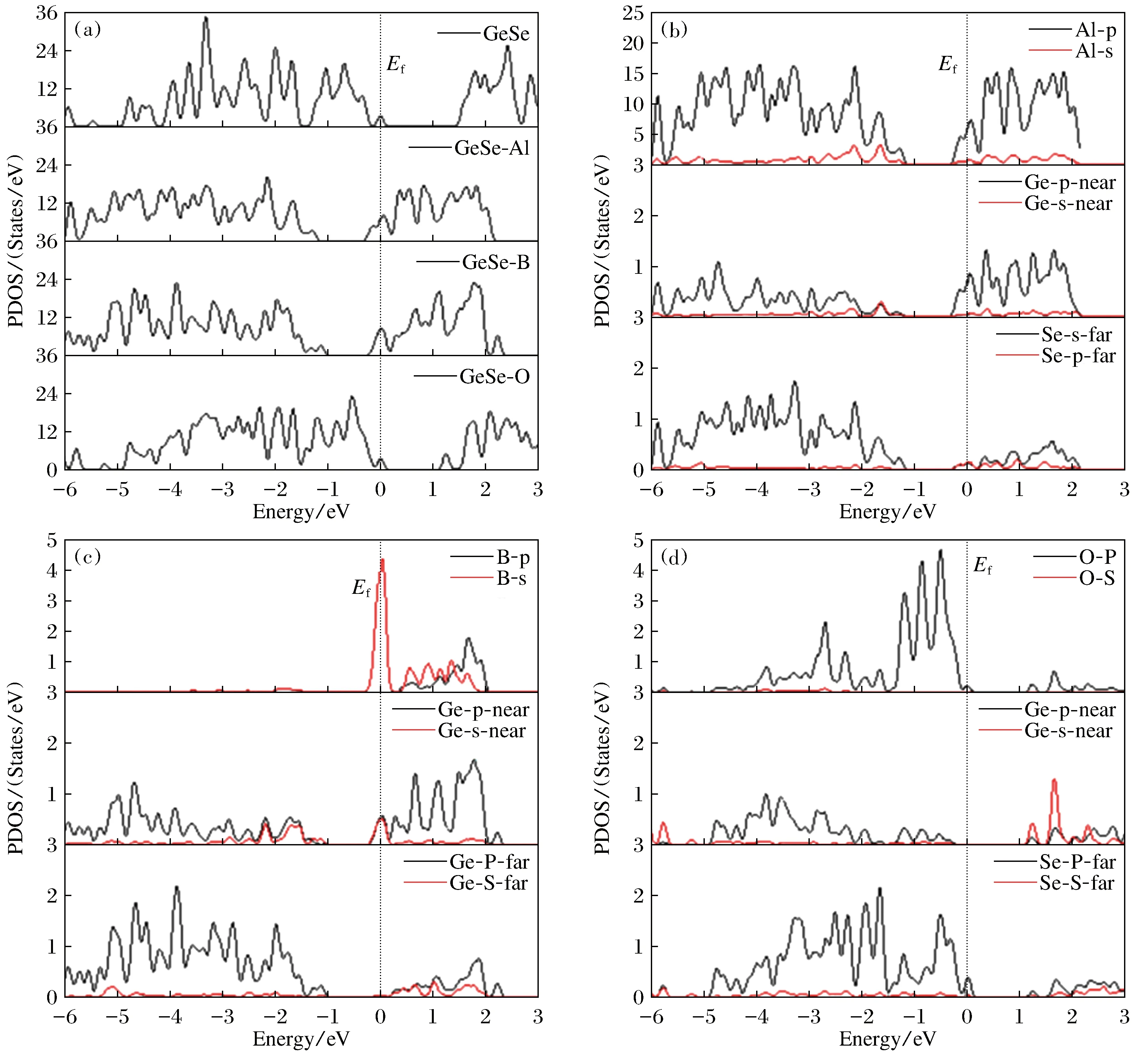
图4 (a) GeSe单层的总态密度图; (b) GeSe单层体系中吸附Al原子及其近邻的Ge,Se原子的分波态密度; (c) GeSe单层体系中吸附B原子及其近邻的Ge, Se原子的分波态密度; (d) GeSe单层体系中吸附O原子及其近邻的Ge, Se原子的分波态密度
图6为GeSe单层在吸附不同原子反射率时随能量的变化曲线。吸附B,O和Al原子以及未吸附原子的GeSe单层静态时反射率分别为0.18,0.18,0.47和0.18,由此可见,吸附Al原子的GeSe单层静态反射率较大,约为47%。吸附B,O与纯净的GeSe单层的反射率近似,约为18%,可见B, O吸附对静态GeSe单层反射太阳光影响不大。随着能量的增加,单层GeSe反射率增大,在能量为7.50 eV时有最大峰值,约为70%,然后随能量的增加,反射率迅速减少。GeSe的反射光谱区域集中于紫外光范围内(>3.10 eV)。由图6可以看出,除了吸附Al原子的GeSe单层的低能区反射率略高,吸附B原子和O原子与未吸附的GeSe在红外光范围(<1.60 eV)和可见光内(1.6~3.1 eV)的反射率相对来说较小,所以GeSe也可作为良好的防紫外线材料,并且可以通过吸附B原子和Al原子来改善材料对紫外线的防护效果。
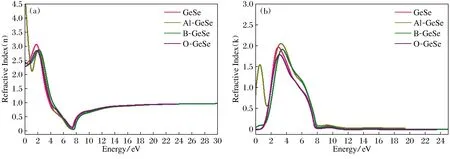
图5 (a)吸附不同原子的GeSe单层的折射率(折射函数实部); (b)吸附不同原子GeSe单层的消光系数(折射函数虚部)
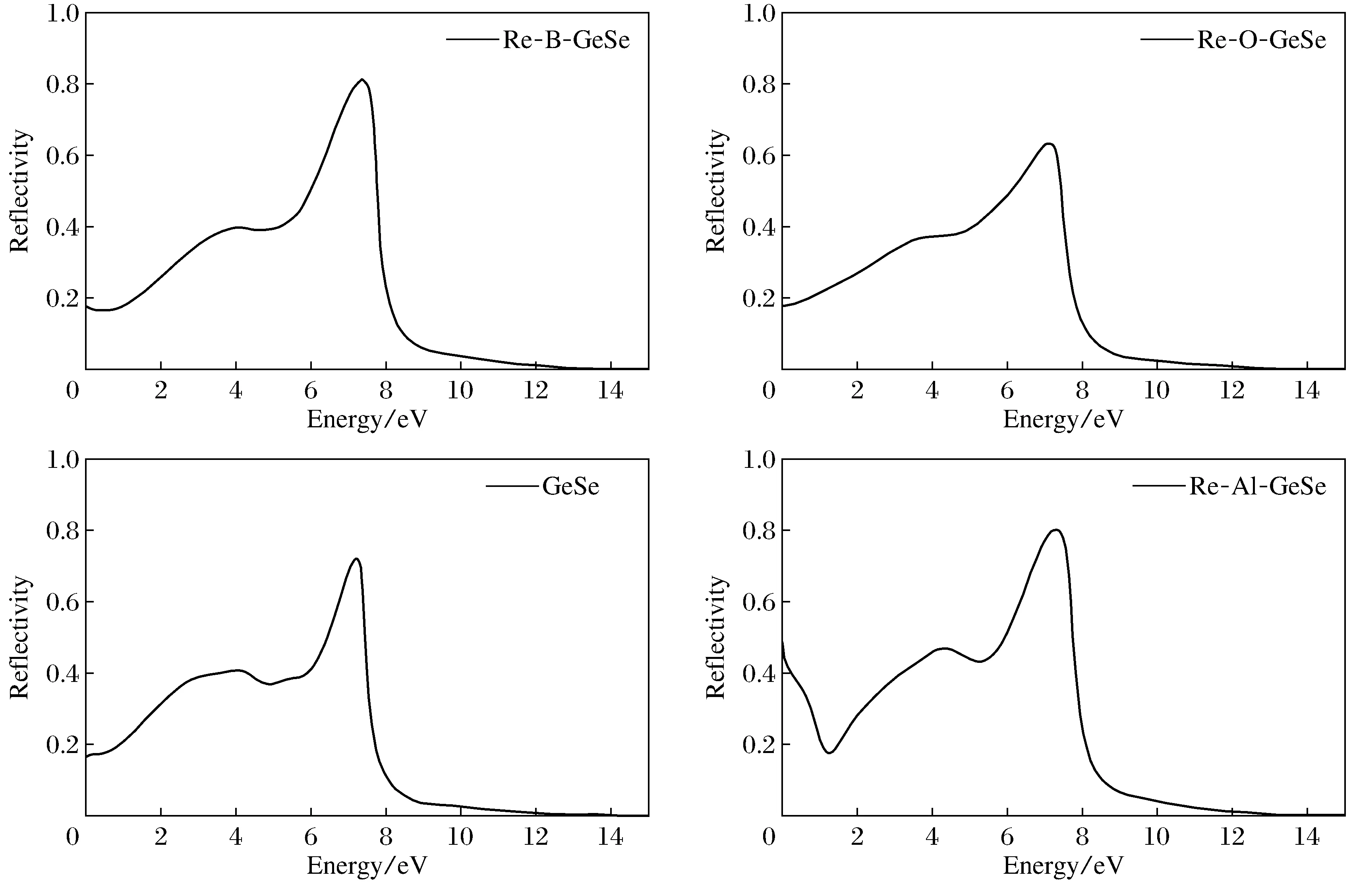
图6 吸附不同原子的GeSe单层的反射率Fig.6 Reflectivity of GeSe single layer absorbed different atom
图7为GeSe单层吸附不同原子时吸收系数随能量的变化曲线图。从图7(a)可以看出,起始吸收限在1 eV左右,与前面能隙计算一致(1.213 eV)。GeSe单层在能量3.00~7.00 eV,有较大的吸收,吸收率大于80 000 cm-1。根据前面态密度图,这些光子能量归因于Se,Ge的p(-2 eV~-6 eV)电子从已占据价带激发到未占据的导带产生的吸收。9.5 eV光子能量的吸收来源于态密度图-8.5 eV左右的价带激发到未占据导带产生的吸收。在能量为3.5 eV处的吸收谱峰值为1.0×105cm-1,对应-4.5 eV价带激发到未占据导带产生的吸收。吸附O原子的GeSe单层的吸收谱线和未吸附原子的GeSe单层的最大吸收谱类似,曲线走势也较为相近。只是最低的吸收限向低能区有所延伸,这与吸附O原子的GeSe单层能隙有所减小有关。O吸附也使得3.00~7.00 eV的吸收有所降低。从整体来看,吸附B原子和Al原子的GeSe单层在能量3.00~7.00 eV的吸收明显增强。同时吸附B原子和Al原子的GeSe单层对光的吸收范围变宽,主要是延伸到红外光区,尤其是吸附Al,在1 eV附近产生了一个吸收峰,这与能带图中-1.0 eV价带顶的电子激发跃迁到费米能级处的杂质能级上对应。可见光的范围是380~780 nm,可见光光子的能量在1.61~3.10 eV。观察吸收谱发现GeSe单层的吸收范围涵盖了从红外到可见光再到紫外的光谱区。
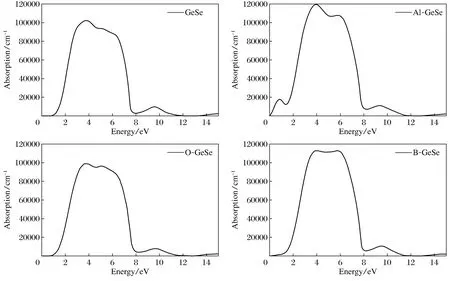
图7 吸附不同原子GeSe单层的吸收系数Fig.7 Adsorption coefficient of GeSe single layer absorbed different atom
3 结 语
本文采用基于密度泛函理论的第一性原理下的GGA算法对GeSe单层的光电性质进行了研究。研究发现交换关联势采用GGA框架下PW91形式较PBE和PBESOL能更好地描述GeSe单层的性质。吸附能计算结果表明,O,Al或 B原子都能在GeSe单层形成稳定的吸附,其中O形成的吸附最稳定。电子结构计算表明,纯净的GeSe单层为直接带隙半导体,原子吸附调制了带隙,同时影响了GeSe单层光吸收的吸收边。能带结构及态密度分析表明,B,Al吸附在能隙中靠近导带底引入了未占据的杂质态,使GeSe单层表现为n型半导体和导体(带隙为0);O吸附引入的杂质带接近价带顶,因而导致GeSe单层为p型半导体。光学性质的计算结果表明,Al吸附GeSe单层在带边附近吸收系数大于未掺杂GeSe单层,显示出通过Al吸附能够有效提高GeSe单层在红外光区光的吸收能力。此外,B,Al吸附都增强了可见光和紫外光区的吸收。反射率结果表明,GeSe单层的反射光谱集中于紫外光范围,B,Al吸附使GeSe单层的反射增强,可见这一吸附可在紫外光的探测和防护上取得较好应用。 同时,折射率和消光系数的计算结果表明,吸附原子都可以对GeSe单层的光学性质进行调制。总之,本研究为GeSe二维材料在光电领域的应用提供了可靠的理论依据。
猜你喜欢
吉首大学学报(自然科学版)(2023年6期)2023-12-22 08:18:20
济南大学学报(自然科学版)(2021年6期)2021-11-10 01:25:42
装备维修技术(2021年36期)2021-10-25 13:21:04
弹箭与制导学报(2021年3期)2021-07-30 02:56:52
成都信息工程大学学报(2019年3期)2019-09-25 08:31:14
网印工业(2019年4期)2019-05-21 06:41:58
重型机械(2019年2期)2019-04-28 11:52:04
吉首大学学报(自然科学版)(2018年3期)2018-07-03 03:14:12
振动工程学报(2017年4期)2018-05-31 12:38:00
电子制作(2018年1期)2018-04-04 01:48:38
