
温压条件下不同含水高岭石对NH4+吸附的分子模拟
2022-08-24 02:06杨有威王有霖罗玉霞刘新棋郭昌胜王春英
中国环境科学 2022年8期
杨有威,王有霖,罗玉霞,刘新棋,郭昌胜,陈 明,王春英*
温压条件下不同含水高岭石对NH4+吸附的分子模拟
杨有威1,3,王有霖2,罗玉霞1,刘新棋1,郭昌胜3,陈 明1,王春英1*
(1.江西省矿冶环境污染控制重点实验室,江西 赣州 341000;2.中国南方稀土集团有限公司,江西 赣州 341000;3.中国环境科学研究院,环境基准与风险评估国家重点实验室,北京 100012)
为研究高岭石对NH4+吸附的微观情况,通过Material Studio软件对高岭石单胞进行收敛性测试后构建了4×2×1不同水化程度高岭石模型,采用量子力学和经典力学方法对模型晶胞进行了理论计算和吸附实验研究.结果显示,在交换关联泛函GGA-PW91,K点4×3×2,截断能600eV条件下,得到了高岭石稳定结构模型(误差<2%);高岭石对NH4+的吸附受温度影响明显,随温度升高,吸附量逐渐减少,与吸附实验结果一致;动力学结果显示吸附类型主要为物理吸附,吸附作用力为范德华力和库仑力.
高岭石;分子模拟;吸附;NH4+;影响因素
稀土资源有着“工业味精”之称,是无法再生的战略性资源.我国南方地区的离子型稀土矿蕴含着中国特有、世上罕有的稀土资源,其中更以存量稀少的中、重稀土为主[1],这些稀土主要以离子形式吸附在黏土矿物表面[2-3].稀土矿一般使用堆浸、池浸或原地浸矿的工艺进行开采,早年使用氯化钠溶液作为浸取剂,但由于浸取率低下等问题改用硫酸铵,既提高了稀土离子的浸出效率,又减少了浸出剂使用量,曾广泛应用于稀土矿开采[4].硫酸铵溶液作为浸取剂开采稀土的过程中,NH4+和黏土矿物表面发生反应,赋存在矿物里的稀土离子通过置换吸附被交换下来[5].
高岭石作为高岭土的主要成分,得名于江西省景德镇的高岭山.高岭石大多呈灰白色或白色,具有土状光泽,分子式为Al4[Si4O10](OH)8,由39.5%AlO3, 46.54%SiO2,13.96%H2O组成,属三斜晶系或单斜晶系.晶体结构单位层是由1个硅氧面(SiO4)和铝氧面(AlO6)构成的1:1型层状结构[6],依靠与O原子连接构成内部结构,单位片层O原子与铝氧面形成氢键,从而构成了重叠的高岭石层状分子[7].高岭石作为离子型稀土矿床中黏土矿物的主要成分之一,研究其特性对稀土资源开发利用或稀土矿山修复有着重要意义[8].通过计算机分子模拟技术,从微观角度研究物质的实验方法,目前已经得到科研人员的认可,并广泛用于分析物质微观性质.国内外许多学者[9-12]对高岭石进行了研究.左骁遥等[13]借助分子模拟软件,搭建了4种黏土矿物模型,运用蒙特卡罗和分子动力学方法,系统研究了高岭石对二氧化碳的吸附性能和分子扩散情况;Niu等[14]采用蒙特卡罗模拟方法研究了氩气在高岭石表面吸附,根据氩在(0 0 1)表面分布和电位,确定了单层吸附机理和吸附位点;Ma等[15]采用分子动力学模拟方法,系统研究了H2O、CO2、CH4、N2、C8H18和C3F8在高岭石(0 0 1)表面吸附构型、密度分布和吸附能,讨论了不同温度和压力条件下各分子结合能、吸附距离和自由能.用分子模拟方法来研究高岭石吸附NH4+,有利于对高岭石吸附特性补充,对稀土矿山土壤中氨氮污染的修复也具有重要意义.
1 材料与方法
1.1 计算模型的构建及优化
运用Materials Studio软件(以下简称MS)2019版本,引用其数据库中的kaolinite.std作为高岭石初始构型(图1,亦称单胞).矿物晶体结构的差异影响其性能;在理论计算中,矿物晶体结构模型是否合理影响着后期理论计算精确性,因此,搭建一个合理的结构模型对于理论研究来说非常重要.Bish[16]等在1989年就对高岭石进行了粉晶衍射实验并采用Rietveld精修技术进行分析,确认高岭石空间群为P1,并确定了晶胞参数.Bish得出的高岭石结构模型参数是目前使用最广泛可靠[17],本文将其参数定为初始晶胞优化的标准.

图1 高岭石单胞优化
选用MS软件的CASTEP模块对单胞模型进行优化.CASTEP模块可对晶体结构进行几何优化,并且能对晶体电子结构或表面性质等进行计算,其中对晶胞结构有主要影响的参数为交换关联泛函、K点和截断能,通过收敛性测试(表1)确定三者参数,以优化后晶格常数相差在2%以内为标准实验值[18].

表1 收敛性测试设置
注:3个因素多个水平共84种组合.
通过收敛性测试来选择交换关联泛函、K点和截断能.根据收敛测试结果与标准值的误差最终选定优化条件为:交换关联泛函选择GGA-PW91,K点选择4´3´2,截断能选择600eV,优化后晶格参数和晶格角度见表2,获得了误差为1.46%(<2%)高岭石稳定结构模型.

表2 最优收敛性测试结果
在获得高岭石稳定结构模型后,考虑到高岭石晶体的周期性和对称性,建立一个4×2×1超胞结构模型.有研究指出,高岭石晶体矿物易于沿(0 0 1)晶面解离,且在(0 0 1)面发生的解离只断裂了结构单元层之间的氢键,在自然破碎下的解离相较于其他晶面更加完全,宏观上高岭石(0 0 1)面面积也是最大的[19].为了验证这一结论,通过MS的Morphology Calculation模块BFDH任务对优化后的高岭石各晶面进行测试.结果如表3所示,(0 0 1)面面积是所有晶面最大的.因此,结合高岭石的特点,运行Build surfaces程序,在cleavesurface中截取(0 0 1)面,截取厚度数值设置为1.0.为了保证周期性边界条件和高岭石真实性,计算模型由1:1层状结构体组成,由于高岭石层间距为0.72nm,因此真空层设置为7.2Å,最后通过Build layer命令得到计算模型[20-21](图2a).

表3 高岭石晶面测试面积
为了获得更优、更稳定的吸附构型,对构建好的高岭石超胞用Forcite模块[22]进行结构优化.优化过程中主要进行以下设置:整个过程高岭石构型保持刚性,晶胞固定非键截断半径设置为9.5Å (设置标准为不超过晶胞最短距离的一半),最大迭代步数设置106,电荷计算选择电荷平衡法,静电作用产生的静电能采用Ewald加和法,范德华力作用选择Atom Based法,力场选择适用于黏土矿物体系的Clayff力场[23].首先通过Steepest descent方法初步优化;为了消除高岭石体系中的重叠构象改用Quasi- newton方法再次对分子模型进行优化,得到作为最终吸附构型用于后续计算的构型图2b.优化前后各能量如表4所示,优化后体系总能量出现大幅下降表明高岭石模型趋向于稳定[24].由于高岭土表面等电点为pH=7.3±0.2[25],在中性时对NH4+的吸附最好,因此文中模型未赋予正/负电荷.

图2 高岭石超胞优化
1.2 吸附模型计算方法
吸附理论计算采用巨正则蒙特卡罗法模拟.由MS软件Sorption模块完成,计算条件为:选择fixed pressure任务,采用Metropolis方法,设置温度为283~363K、压力为101.32kPa,勾选返回5个能量构型,使用黏土矿物Clayff力场,电荷计算适用力场电荷(Forcefield assigned),其他条件同优化条件一致,吸附质NH4+使用Assign手动分配电荷.选择返回的最低能量,在Forcite模块下选择分子动力学(Dynamics)任务,采用正则系综进行动力学模拟.模拟时间步长设置成1fs,时间1000ps,总步数为106,力场使用Universal力场,电荷计算使用本身电荷(Use current),静电能选择Ewald加和法,范德华力作用选择Atom Based法,其余条件参数同吸附模拟参数.

表4 高岭石模型优化前后能量变化(kJ/mol)
实际条件下水的存在不可避免,水分子会进入晶胞内产生作用力(水化作用),无水环境吸附过于理想,因此需要对高岭石吸附构型加入不同质量比例的水分子模拟水环境(以下简称%水模型,=0,6, 12,24).水环境构建通过Adsorption模块Simulated annealing命令实现,如图2c所示,水分子加入后同样进行计算,各条件参数不变.
1.3 蒙特卡罗模拟
计算直接获得的NH4+吸附量是每个晶胞内NH4+吸附个数,单位转换公式(1)[26]如下:

式中:代表晶胞密度,g/cm3;代表阿伏伽德罗常数,6.02´1023;代表晶胞体积,cm3;ab为绝对吸附量,mmol/g.
单位转化后得到的值为绝对吸附量,是超临界吸附[27],不能全部视为吸附量,因此,需要利用公式(2)[28]将绝对吸附量转化为超额吸附量,超额吸附量即最终结果:

式中:g为气相密度,g/cm3;a为吸附相体积,g/cm3;ab为绝对吸附量,mmol/g;ex为超额吸附量,mmol/g.
PR方程可解得气相密度,a可用自由体积p来表示,使用MS软件中Tools选项中的Atom Volumes & Surface 模块可以得到晶胞的自由体积p.
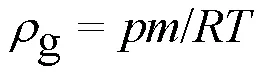
式中:为压强,kPa;为质量,g;为常数8.314;为温度,K.

式中:为NH4+吸附量,mg/kg,为NH4+的相对分子质量,18.
1.4 NH4+在高岭石表面的吸附
首先配制40mmol/L的硫酸铵(西陇科技股份有限公司,AR分析纯)溶液,然后,秤取4g高岭土(国药集团化学试剂有限公司,AR分析纯)于100mL三角锥瓶中,加入20mL硫酸铵溶液,将三角锥瓶置于恒温磁力搅拌器(常州金坛良友仪器有限公司, HJ-A6型)上,控制温度恒温磁力搅拌4h使其达到吸附平衡.试样6000r/min离心分离5min后将固体物质置于真空干燥箱中干燥,按重量法[29]计算高岭土饱和含水率,同时取上清液过0.45μm滤膜后按照纳氏试剂分光光度法[30]使用紫外可见分光光度计(上海元析仪器有限公司,B-800型)对NH4+定量分析, NH4+吸附量计算公式见式(5):
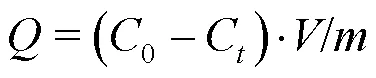
式中:为试样容量,mL;为试样质量,g;0为初始NH4+浓度,mg/kg;为吸附平衡后NH4+浓度,mg/kg.
2 结果与讨论
2.1 吸附量
303K条件下高岭石对NH4+的吸附构型见图3.由图4可知,0%水模型下对NH4+的吸附量最大, 283K吸附量为660.4450mg/kg,且吸附量随着温度升高而逐渐减小,其原因是温度升高使得吸附体系熵值和混乱度增加,使得NH4+接触高岭石表面机会增大,但温度升高赋予了NH4+较多动能,NH4+与高岭石之间的吸附强度变弱,NH4+更容易脱离高岭石束缚,最终解吸量NH4+大于被吸附量,造成吸附量降低[31].
杨帅[32]利用以高岭石为主要黏土矿物的南方离子型稀土矿解吸氨氮,在实验中以浸提方式测得了298K不同吸附态氨氮的含量,最大饱和吸附平衡后物理态吸附氨氮含量为254.1000mg/kg(转换为NH4+含量约为326.6067mg/kg),与303K 6%水模型结果相近.

图3 303K条件下高岭石对NH4+的吸附构型
由于理论计算的高岭石矿物模型为单一矿物,实际样品成分多样,可能存在不发生吸附的杂质,因此理论计算结果与实际有一定差异.宋晨曦等[33]在对高岭石吸附解吸氨氮实验中发现物理吸附态氨氮通过范德华力吸附在高岭土表面,化学吸附态氨氮以共价键的形式被高岭土固定.由图4知,随着水分子比例增加,NH4+吸附量减小,说明水分子存在影响了范德华力或共价键,部分NH4+的活性位点被水分子占据,使得NH4+吸附量小于0%水模型.此外,0%和6%水模型吸附量受温度的影响较大,24%水模型受影响最小.

图4 不同温度下NH4+的吸附量
2.2 等量吸附热
物质吸附条件(、和理化性质)不变时,一定量(1mol)吸附质被吸附时产生的焓变即为等量吸附热[34].等量吸附热与吸附量之间关系复杂多变,它可随着吸附量变化而变化或不变,但与吸附剂表面理化结构有关,熊健等[35]的研究指出等量吸附热与吸附剂孔径大小变化有关,等量吸附热随着蒙脱石狭缝孔径的增大而减小,说明吸附会由能量较低的吸附位点向能量较高的位点转移.
图5中4种水模型吸附NH4+等量吸附热为9.5~ 13.0kJ/mol,均小于42kJ/mol.研究表明[36],等量吸附热小于42kJ/mol的吸附类型为物理吸附,反之为化学吸附,因此4种水模型吸附主要吸附类型为物理吸附,吸附可能靠分子间作用力实现,无化学键断裂或生成.此外,等量吸附热随温度升高或水比例增加而增加,表明高岭石表面能量呈不均匀分布[37],高岭石不同吸附时期各吸附位点的NH4+吸附势能不同,水分子的存在可能改变了高岭石表面理化性质.吸附初期, NH4+优先向高岭石的高能吸附位点移动,这时吸附所需能量不多,产生的等量吸附热最大,随着吸附过程不断进行, NH4+吸附量逐渐增加,高能吸附位点饱和后吸附转向低能吸附位点,此时吸附所需能量变多,等量吸附热减少,因此等量吸附热随着吸附量的降低表现出升高的趋势,与李晓媛等[38]实验结论一致,水分子增加比温度升高导致等量吸附热升高的趋势更加明显.
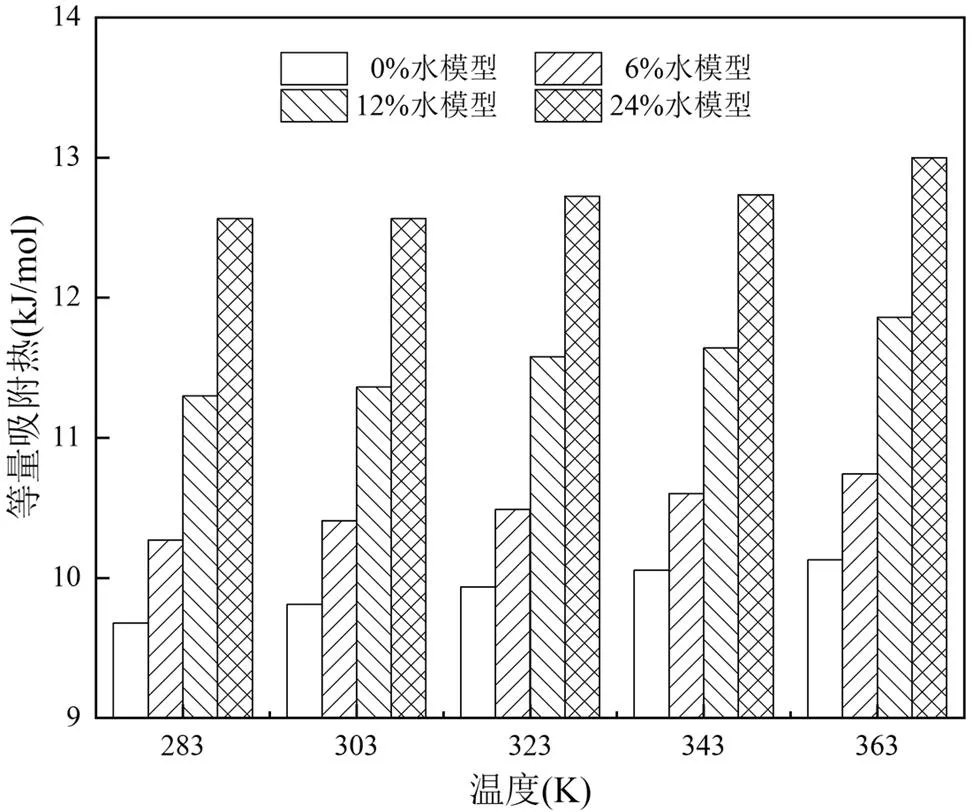
图5 不同温度下吸附NH4+的等量吸附热
2.3 分子动力学模拟
2.3.1 浓度分布 为了进一步分析NH4+的分布情况,在高岭石(0 0 1)面C轴方向(即垂直方向)分析吸附浓度分布.图6是经过Forcite模块分子动力学模拟返回的计算结果,可以看出,经过分子动力学模拟,不同条件下的NH4+分布分为两个吸收峰,峰高相似,主要吸收峰不明显,具有一定的对称性,峰1出现在5~7Å,峰2出现在13~17Å,考虑高岭石层间距并结合结构周期性得知峰1为铝氧层,峰2为硅氧层,还有少部分零散分布在两峰之间的游离吸附层.对比图6发现,随着水分子加入的增多,高岭石吸附浓度在逐渐降低,说明水分子存在影响了分子间作用力,与图4的结果一致.
2.3.2 径向分布函数 4种水模型对NH4+的吸附作用力尚不确定,确定二者间作用力可通过径向分布函数进行分析[39].以某个粒子半径为圆心,描述出现其他粒子的概率是径向分布函数的核心含义,求解如公式(6).

式中:n为半径r范围内的其他粒子数;ρ为晶体密度,g/cm3;N为半径r范围内的其他粒子数;g(r)为径向分布函数.
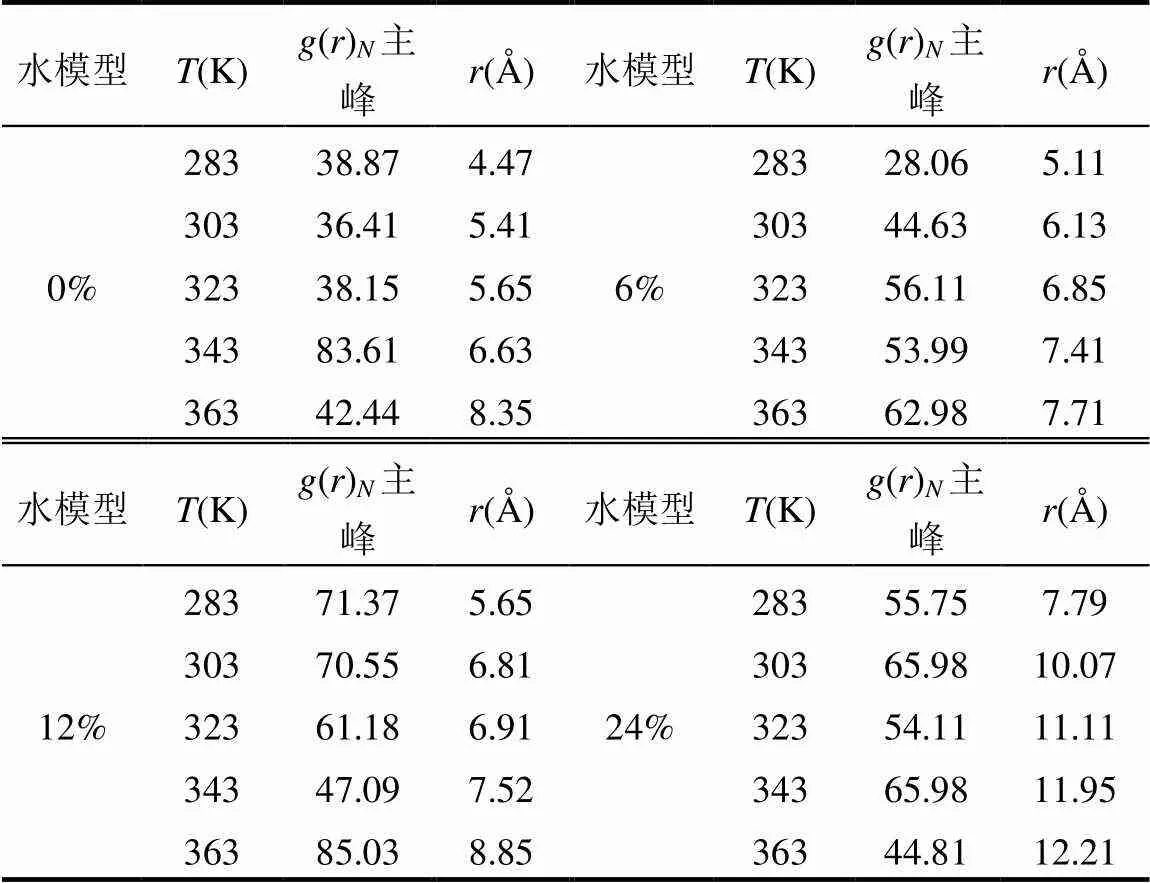
表5 径向分布函数峰值g(r)和所在半径r
注:()指以为圆心的径向分布函数.
径向分布函数结果,如果峰所对应的半径小于或等于3.5Å,说明固定离子和研究对象离子之间产生了化学键或氢键;如果峰值所对应的半径大于3.5Å,说明固定离子和研究对象之间靠库仑力和范德华力维持[40].分子动力学计算完成后可通过Forcite模块中的Radial distribution function得到(),结果如表5,4种水模型()峰值对应的半径随着温度升高而增大,说明温度升高各粒子活跃度增加,并促使()峰值出现在较远位置,而峰值所对应的半径均在3.5Å以外,因此高岭石对NH4+的吸附作用力为范德华力和库仑力.
2.4 NH4+在高岭石表面吸附结果
由图7可知,随着吸附反应温度升高,实验值和模拟值均降低,各温度高岭土饱和含水率为10%~ 13%,与12%水模型相近,但实验值和模拟值有偏差.这是因为理想化模型与实际样品有一定差别,如层间阳离子、原子类质同相替代位置不同等[41].据报道,高岭土受温度影响,在低温时颗粒间结合力最弱[42],且NH4+在温度较低时从固相逃逸的趋势最低[43],理论模型因为真空层的关系受温度影响较小,因此283K时实验值和模拟值的差异较其他温度大.
吸附实验显示吸附量随温度升高而减少,与理论计算结果一致.温度升高使吸附体系中分子混乱度增加,NH4+容易获得突破作用力吸附的动能,造成被吸附固定下来的NH4+变少[44];另有李贞[45]的研究结果显示,高岭土吸附NH4+过程为放热反应,硅氧面和铝氧面吸附能分别为-53.07,-117.81kJ/mol,温度升高不利于放热反应,因此降低吸附量.

图7 高岭土对NH4+吸附的实验值、模拟值和饱和含水率
3 结论
3.1 通过MS分子模拟工具,利用量子力学方法优化减小误差后用Build模块构建了4种不同水化程度的高岭石(0 0 1)表面分子模型,并经过Forcite模块优化得到了高岭石稳定吸附构型.
3.2 蒙特卡罗模拟结果显示,吸附温度和水化程度对高岭石吸附NH4+具有抑制作用,吸附量随温度升高而降低,水化程度越深高岭石越难吸附NH4+;吸附实验结果表明,高岭石饱和含水率与12%水模型相近,吸附NH4+的量随温度的升高表现出降低的趋势,符合理论计算结果.
3.3 动力学模拟浓度分布曲线表明高岭石吸收峰分为硅氧面和铝氧面,水分子加入会影响分子间作用力,使得硅氧面和铝氧面吸附量减少;径向分布函数峰值所对应的半径均大于3.5Å,高岭石与NH4+的吸附作用力为范德华力和库仑力.
[1] 谢芳芳,王观石,罗嗣海,等.离子型稀土尾矿残留铵缓释性分析 [J]. 中国环境科学, 2021,41(9):4333-4340.
Xie F F, Wang G S, Luo S H, et al. Sustained release analysis of residual ammonium from ionic rare earth tailings [J]. Chinese Environmental Science, 2021,41(9):4333-4340.
[2] Huang X W, Long Z Q, Wang L S, et al. Technology development for rare earth cleaner hydrometallurgy in China [J]. Rare Metals, 2015, 34(4):215-222.
[3] Xiao Y F, Feng Z Y, Hu G H, et al. Leaching and mass transfer characteristics of elements from ion-adsorption type rare earth ore [J]. Rare Metals, 2015,34(5):357-365.
[4] 李 琪,秦 磊,王观石,等.离子吸附型稀土浸矿机制研究现状 [J]. 中国稀土学报, 2021,39(4):543-554.
Li Q, Qin L, Wang G S, et al. Research status of ion adsorption type rare earth leaching mechanism [J]. Chinese Journal of Rare Earth sciences, 2021,39(4):543-554.
[5] 赖 城,周 豪,张大超,等.重稀土元素钇对短程反硝化工艺的影响 [J]. 中国环境科学, 2021,41(7):3221-3228.
Lai C, Zhou H, Zhang D C, et al. Effect of heavy rare earth element yttrium on short-cut denitrification process [J]. Chinese Environmental Science, 2021,41(7):3221-3228.
[6] 王 鹏,林雪玲,潘凤春,等.模拟计算压力对高岭石结构与力学性能的影响 [J]. 硅酸盐学报, 2018,46(12):1788-1794.
Wang P, Lin X L, Pan F C, et al. The influence of pressure on structure and mechanical properties of kaolinite was simulated [J]. Journal of silicates, 2018,46(12):1788-1794.
[7] White C E, Provis J L, Riley D P, et al. What is the structure of kaolinite? Reconciling theory and experiment [J]. The Journal of Physical Chemistry B, 2009,113(19):6756-6765.
[8] 邓振乡,秦 磊,王观石,等.离子型稀土矿山氨氮污染及其治理研究进展 [J]. 稀土, 2019,40(2):120-129.
Deng Z X, Qin L, Wang G S, et al. Research progress on ammonia nitrogen pollution and its treatment in ionic rare earth mines [J]. Rare earth, 2019,40(2):120-129.
[9] 牛广欣,张 彬,康天和,等.孔隙压力和含水量对煤系高岭石吸附甲烷能量影响的分子模拟研究 [J]. 采矿与安全工程学报, 2018,35(6): 1269-1276.
Niu G X, Zhang B, Kang T H, et al. Molecular simulation of the effect of pore pressure and water content on methane adsorption energy of kaolinite in coal measures [J]. Journal of Mining and Safety Engineering, 2018,35(6):1269-1276.
[10] Ma Y, Lu G, Shao C, et al. Molecular dynamics simulation of hydrocarbon molecule adsorption on kaolinite (0 0 1) surface [J]. Fuel, 2019,237(2):989-1002.
[11] Han Y, Liu W, Chen J. DFT simulation of the adsorption of sodium silicate species on kaolinite surfaces [J]. Applied Surface Science, 2016,370(5):403-409.
[12] Kong X P, Wang J. Copper (II) adsorption on the kaolinite (0 0 1) surface: Insights from first-principles calculations and molecular dynamics simulations [J]. Applied Surface Science, 2016,389(12):316-323.
[13] 左骁遥,房晓红,曾凡桂.二氧化碳在高岭石孔隙中吸附的分子模拟 [J]. 矿产综合利用, 2020,41(1):163-167.
Zuo X Y, Fang X H, Zeng F G. Molecular simulation of carbon dioxide adsorption in kaolinite pores [J]. Comprehensive utilization of mineral resources, 2020,41(1):163-167.
[14] Niu J, Wang D, Wu A, et al. Molecular simulation study of argon adsorption on kaolinite surface with an experimental comparison [J]. Applied Surface Science, 2019,478(6):230-236.
[15] Ma Y, Lu G, Shao C, et al. Molecular dynamics simulation of hydrocarbon molecule adsorption on kaolinite (0 0 1) surface [J]. Fuel, 2019,237(2):989-1002.
[16] Bish D L. Rietveld refinement of the kaolinite structure at 1.5K [J]. Clays and clay minerals, 1993,41(6):738-744.
[17] 吕文婷,程 运,谢 浩,等.基于密度泛函理论的高岭石吸附机理研究进展 [J]. 硅酸盐通报, 2020,39(1):157-168.
Lv W T, Cheng Y, Xie H, et al. Research progress on adsorption mechanism of kaolinite based on density Functional Theory [J]. Silicate Bulletin, 2020,39(1):157-168.
[18] 陈 军,闵凡飞,刘令云,等.不同胺/铵阳离子在高岭石(0 0 1)面吸附的密度泛函计算 [J]. 煤炭学报, 2016,41(12):3115-3121.
Chen J, Min F F, Liu L Y, et al. Different amine/ammonium cations in kaolinite (0 0 1) surface adsorption density functional calculations [J]. Journal of coal, 2016,41(12):3115-3121.
[19] 戴 伟,水中和,沈春华,等.水分子在高岭土中吸附特性的蒙特卡罗模拟研究 [J]. 硅酸盐学报, 2012,40(1):149-153.
Dai W, Shui Z H, Shen C H, et al. Monte Carlo simulation of adsorption characteristics of water molecules in kaolin [J]. Journal of the Chinese Ceramic Society, 2012,40(1):149-153.
[20] 傅梁杰,刘天宇,杨华明.黏土矿物材料表界面功能设计的计算模拟 [J]. 硅酸盐学报, 2021,49(7):1347-1358.
Fu L J, Liu T Y, Yang H M. Computational simulation of interface functional design of clay mineral materials [J]. Journal of the Chinese Ceramic Society, 2021,49(7):1347-1358.
[21] Santana E, Possa R D, Novais A L F, et al. Adsorption study of 4-nitrophenol onto kaolinite (001) surface: A van der Waals density functional study [J]. Materials Chemistry and Physics, 2021,271:124887.
[22] Akkermans R L C, Spenley N A, Robertson S H. Monte carlo methods in materials studio [J]. Molecular Simulation, 2013,39(14/15):1153-1164.
[23] 张亚云,陈 勉,邓 亚,等.温压条件下蒙脱石水化的分子动力学模拟 [J]. 硅酸盐学报, 2018,46(10):1489-1498.
Zhang Y Y, Chen M, Deng Y, et al. Molecular dynamics simulation of montmorillonite hydration under temperature and pressure [J]. Journal of silicates, 2018,46(10):1489-1498.
[24] Dubbeldam D, Torres-Knoop A, Walton K S. Monte Carlo codes, tools and algorithms [J]. Molecular Simulation, 2013,39(14/15):1253-1292.
[25] Rand B, Melton I E. Particle interactions in aqueous kaolinite suspensions: I. Effect of pH and electrolyte upon the mode of particle interaction in homoionic sodium kaolinite suspensions [J]. Journal of Colloid and Interface Science, 1977,60(2):308-320.
[26] 张凯飞.煤层气排采过程中甲烷解吸与扩散过程的分子模拟研究 [D]. 太原:中北大学, 2020.
Zhang K F. Molecular simulation of methane desorption and diffusion during coalbed methane drainage [D]. Taiyuan: North University of China, 2020.
[27] Guo F, Wang S, Feng Q, et al. Adsorption and absorption of supercritical methane within shale kerogen slit [J]. Journal of Molecular Liquids, 2020,320(11):43-64.
[28] 熊 健,刘向君,梁利喜.甲烷在蒙脱石狭缝孔中吸附行为的分子模拟 [J]. 石油学报, 2016,37(8):1021-1029.
Xiong J, Liu X J, Liang L X. Molecular simulation of methane adsorption in montmorillonite slit pores [J]. Acta Petrolei Sinica, 2016,37(8):1021-1029.
[29] HJ613-2011 土壤干物质和水分的测定重量法 [S].
HJ613-2011 Soil - Determination of dry matter and moisture - gravimetric method [S].
[30] HJ 535-2009 水质氨氮的测定纳氏试剂分光光度法 [S].
HJ 535-2009 Determination of water quality and ammonia nitrogen by Spectrophotometry with Nahler's reagent [S].
[31] 杨 飞,房晓红,曾凡桂,等.高岭石表面吸附铅和镉的模拟计算 [J]. 矿产综合利用, 2020,41(5):196-202,100.
Yang F, Fang X H, Zeng F G, et al. Simulation of Pb and Cd adsorption on kaolinite surface [J]. Multipurpose Utilization of Mineral Resources, 2020,41(5):196-202,100.
[32] 杨 帅.离子型稀土矿开采过程中氨氮吸附解吸行为研究 [D]. 北京:中国地质大学(北京), 2015.
Yang S. Adsorption and desorption behavior of ammonia nitrogen in ionic rare earth ore mining [D]. Beijing: China University of Geosciences (Beijing), 2015.
[33] 宋晨曦,秦 磊,胡世丽,等.离子型稀土尾矿除铵效果对比 [J]. 环境工程学报, 2019,13(4):969-976.
Song C X, Qin L, Hu S L, et al. Comparison of ammonium removal from ionic rare earth tailings [J]. Chinese Journal of Environmental Engineering, 2019,13(4):969-976.
[34] 郭 为,熊 伟,高树生,等.温度对页岩等温吸附/解吸特征影响 [J]. 石油勘探与开发, 2013,40(4):481-485.
Guo W, Xiong W, Gao S S, et al. Effect of temperature on isothermal adsorption/desorption characteristics of shale [J]. Petroleum Exploration and Development, 2013,40(4):481-485.
[35] 熊 健,刘向君,梁利喜.甲烷在黏土矿物狭缝孔中吸附的分子模拟研究 [J]. 煤炭学报, 2017,42(4):959-968.
Xiong J, Liu X J, Liang L X. Molecular simulation study of methane adsorption in clay mineral slit pores [J]. Journal of China Coal Society, 2017,42(4):959-968.
[36] 傅献彩,沈文霞,姚天扬.物理化学(第四版)上册 [M]. 北京:高等教育出版社, 1990.
Fu X C, Shen W X, Yao T Y. Physical chemistry (4th Ed.) vol. I [M]. Beijing: Higher Education Press, 1990.
[37] 相建华,曾凡桂,梁虎珍,等.CH4/CO2/H2在煤分子结构中吸附的分子模拟 [J]. 中国科学: 地球科学, 2014,44(7):1418-1428.
Xing J H, Zeng F G Liang H Z, et al. Molecular simulation of CH4/CO2/H2adsorption in coal molecular structure. Science China Earth Sciences, 2014,44(7):1418-1428.
[38] 李晓媛,曹 峰,岳高凡,等.柴达木盆地东部石炭系页岩吸附特性实验研究 [J]. 地学前缘, 2016,23(5):95-102.
Li X Y, Cao F, Yue G F, et al. Experimental study on adsorption characteristics of Carboniferous shales in eastern Qaidam Basin [J]. Earth Science Frontiers, 2016,23(5):95-102.
[39] 金肇岩,胡筱敏,孙 通,等.脉冲电吸附技术深度脱氮及分子动力学模拟 [J]. 中国环境科学, 2019,39(7):2871-2879.
Jin Z Y, Hu X M, Sun T, et al. Deep nitrogen removal and molecular dynamics simulation by pulsed electroadsorption [J]. Chinese Environmental Science, 2019,39(7):2871-2879.
[40] Allen M P, Tildesley D J. Computer simulation of liquids [M]. Oxford university press, 2017.
[41] 孙仁远,张云飞,范坤坤,等.页岩中黏土矿物吸附特性分子模拟 [J]. 化工学报, 2015,66(6):2118-2122.
Sun R Y, Zhang Y F, Fan K K, et al. Molecular simulation of adsorption characteristics of clay minerals in shale [J]. Ciesc Journal, 2015,66(6):2118-2122.
[42] Zhao Y, Zhang B, Zhang X, et al. Preparation of highly ordered cubic NaA zeolite from halloysite mineral for adsorption of ammonium ions [J]. Journal of Hazardous Materials, 2010,178(1-3):658-664.
[43] Mon E E, Hamamoto S, Kawamoto K, et al. Temperature effects on geotechnical properties of kaolin clay: simultaneous measurements of consolidation characteristics, shear stiffness, and permeability using a modified oedometer [J]. GSTF Journal of Geological Sciences, 2013, 1(1):1-10.
[44] Huang S, Feng J, Yu J, et al. Adsorption and desorption performances of ammonium on the weathered crust elution-deposited rare earth ore [J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2021,613:126139.
[45] 李 贞.高岭石表面上铵离子吸附的量子化学研究[J]. 有色金属科学与工程, 2017,8(4):36-41.
Li Z. Quantum chemistry study of ammonium ion adsorption on kaolinite surface [J]. Nonferrous Metals Science and Engineering, 2017,8(4):36-41.
Molecular simulation of NH4+adsorption by kaolinite with different water content under temperature and pressure.
YANG You-wei1,3, WANG You-lin2, LUO Yu-xia1, LIU Xin-qi1, GUO Chang-sheng3, CHEN Ming1, WANG Chun-ying1*
(1.Jiangxi Key Laboratory of Mining and Metallurgy Environmental Pollution Control, Ganzhou 341000, China;2.China South Rare Earth Group Co. LTD, Ganzhou 341000, China;3.State Key Laboratory of Environmental Criteria and Risk Assessment, Chinese Research Academy of Environmental Sciences, Beijing 100012, China)., 2022,42(8):3720~3727
In order to study the microscopic situation of NH4+adsorption by kaolinite, kaolinite models with different hydration degrees of 4×2×1 were constructed after the convergence test of kaolinite monocytes by Material Studio software. The theoretical calculation and adsorption experiment of the model cell were carried out by using the methods of quantum mechanics and classical mechanics. The results showed that the stable structure model of kaolinite is obtained (error < 2%) under the conditions of exchange correlation functional GGA-PW91, K point 4×3×2 and truncation energy 600eV. The adsorption of NH4+by kaolinite was obviously affected by temperature. With the increase of temperature, the adsorption capacity decreases gradually, which was consistent with the experimental results. The kinetic results showed that the adsorption type was mainly physical adsorption, and the adsorption forces were Van der Waals force and Coulomb force. The research results were helpful to supplement the research on the kaolinite properties and have a guiding role in the control of ammonia nitrogen pollution in soil of rare earth mines.
kaolinite;molecular simulation;adsorption;NH4+;influencing factors
X53,TD985
A
1000-6923(2022)08-3720-08
2022-01-10
国家重点研发计划(2019YFC1805100);国家自然科学基金资助项目(21767012);江西理工大学清江青年英才支持计划项目(JXUSTJYX2016003);江西理工大学研究生创新专项资金项目(XY2021-S013)
* 责任作者, 副教授, cywang@jxust.edu.cn
杨有威(1998-),男,广东韶关人,江西理工大学硕士研究生,主要研究方向为分子模拟与水污染控制技术.发表论文1篇.
猜你喜欢
导航定位学报(2022年4期)2022-08-15
疯狂英语·新读写(2021年8期)2021-11-05
中学生数理化·七年级数学人教版(2020年10期)2020-11-26
农村青少年科学探究(2020年5期)2020-08-18
小学生优秀作文(高年级)(2018年4期)2018-09-11
新民周刊(2018年8期)2018-03-02
饮食科学(2017年12期)2018-01-02
汽车导报(2017年5期)2017-08-03
中学生数理化·高二版(2016年4期)2016-05-14
少儿科学周刊·儿童版(2016年1期)2016-03-14
