
衍生化-液质联用法测定噁拉戈利钠中2-氟-3-甲氧基苯硼酸
2022-08-23 13:20:50胡素招钱暮霜
上海医药 2022年15期
胡素招 钱暮霜
(上海奥博生物医药股份有限公司 上海 201203)
基因毒性杂质,是指能够直接或间接(如通过代谢激活之后)与DNA(主要是其碱基)发生化学反应的物质。近年来,陆续从沙坦类、雷尼替丁、二甲双胍等药物中检测出亚硝胺类杂质残留[1-6]。2020年5月,国家药监局也发布了《化学药物中亚硝胺类杂质研究技术指导原则(试行)》[7]指导纲领。基因毒性杂质的控制贯穿药品研制的全生命周期,是一项复杂而精细的工程。
硼酸及硼酸酯已成为有机合成中重要片段,是药物中的关键中间体。2-氟-3-甲氧基苯硼酸在噁拉戈利钠的合成工艺中作为起始物料被使用,采用(定量)构-效关系(QSAR)方法进行计算机模拟的毒性评估,以预测细菌突变试验的结果[8]。通过CASE Ultra构-效模拟软件分析2-氟-3-甲氧基苯硼酸为阳性,在PHARM_BMUT数据库中QSAR计算的概率为57.2%,在GT_EXPERT数据库中QSAR计算的概率为62.1%,因此,该化合物风险评估归为“有警示结构,与原料药结构无关;无致突变性数据”的3类杂质。参考ICH M7[9-10],监管部门把1.5 μg/d的可接受摄入量作为基因毒性杂质的毒理学阈值,药品噁拉戈利钠的口服最大日剂量为400 mg,故需要在中间体、粗品或成品中控制2-氟-3-甲氧基苯硼酸的限度为0.000 38%。通过衍生化-液质联用法来检测噁拉戈利钠中的2-氟-3-甲氧基苯硼酸的含量,采用铵离子螯合和化学衍生法(图1)技术,在流动相体系中添加10 mmol/L的铵离子,采用配备常规ESI源的液质联用(LCMS)单四级杆分析系统,在SIM模式下用m/z 299.1 (299.1为硼酸酯螯合铵离子[M+NH4]+)进行定量分析,灵敏度高、方法简单、结果可靠、重现性好,满足目前法规规定的限量要求。
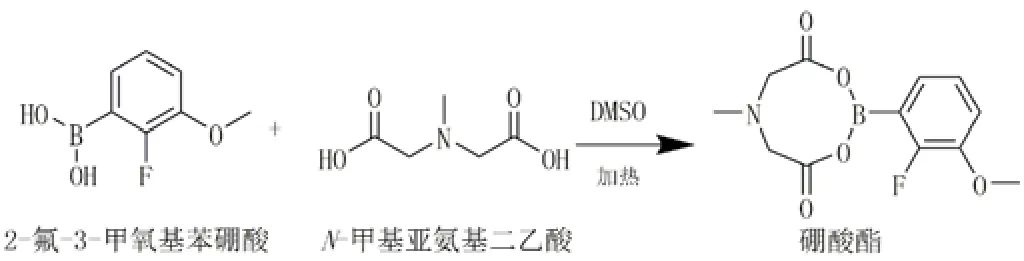
图1 2-氟-3-甲氧基苯硼酸化学结构式和衍生化反应
1 材料和方法
1.1 试剂和样品
乙腈、甲酸、二甲亚砜、乙酸均为色谱级,购于Merck公司;甲酸铵为色谱级,购于CNW公司;N-甲基亚氨基二乙酸为分析级,购于TCI公司;超纯水、2-氟-3-甲氧基苯硼酸对照品和噁拉戈利钠原料药和粗品均由公司自制。
1.2 仪器和设备
Agilent 1260液相色谱仪和Agilent 6120四极杆质谱仪(美国安捷伦公司);XS205DU型电子天平(瑞士梅特勒托利多公司);DHG-9015A鼓风干燥箱(上海一恒科学仪器有限公司)。
1.3 实验方法
1.3.1 色谱和质谱条件
使用Agilent Zorbax Eclipse XDB-C18色谱柱(2.1 mm × 50 mm, 3.5 µm)。采用梯度洗脱模式,甲酸铵水溶液(10 mmol/L,用甲酸调pH 4.0)用作流动相A和乙腈用作流动相B,洗脱时间0 ~ 8 min,B保持 8%;8 ~9 min,B升至95%,再保持3 min,12.0 ~ 12.1 min,B降至8%,再保持3 min。进样体积为3 µL,柱温为40 ℃,流速为0.6 mL/min。
质谱仪在电喷雾电离(ESI)中采用正离子模式,选择离子监测(SIM)模式用于分析物质量检测。典型的质谱参数如下:毛细管电压:3.0 kV,干燥气的流速和温度分别为12.0 L/min和350 ℃,雾化气压力:60 psi,碎片能量:70 V,SIM 离子:299.1 m/z([M+NH4]+为硼酸酯螯合铵离子,质谱采集时间:2.0~9.0 min,峰宽:0.10 min。
1.3.2 溶液配制
1)稀释液 二甲亚砜-乙酸(10∶1)(体积比)。
2)衍生剂 称取250 mgN-甲基亚氨基二乙酸于25 mL容量瓶中(质量浓度10 mg/mL),用稀释液溶解并定容至刻度,混匀,如有不溶物需过滤。
3)空白溶液 准确移取稀释液500 µL和衍生剂500µL于1.5 mL液相小瓶中,混匀,并将混合物于100 ℃加热1 h,之后使温度重新平衡至室温,待测。
4)系列对照品储备溶液 精确称取2-氟-3-甲氧基苯硼酸对照品,加稀释液超声溶解,并定量稀释制成每1 mL中含760 ng的高浓度对照品储备溶液。用稀释液逐级稀释制成每1 mL中分别包含15.2、45.6、76.0、121.6、152.0、182.4、228.0 ng的溶液,作为系列对照品储备溶液;再分别准确移取系列对照品储备溶液500 µL和衍生剂500 µL于1.5 mL液相小瓶中,混匀,并将混合物加热至100 ℃持续1 h,之后使温度重新平衡至室温,分别制成1 mL中分别包含7.6[检出限(LOD),10%水平]、22.8[定量限(LOQ),30%水平]、38.0(50%水平)、60.8(80%水平)、76.0(100%水平)、91.2(120%水平)、114.0(150%水平) ng的硼酸酯溶液,作为系列对照品溶液,待测。用其响应值绘制线性曲线,线性范围和相关浓度点可根据检测需要自行调整。
5)供试品溶液 精密称取400 mg供试品,置于10 mL容量瓶中,用稀释液溶解并定容至刻度,混匀,再准确移取500 µL该溶液和500 µL衍生剂到1.5 mL液相小瓶,混匀,并将混合物加热至100 ℃持续 1 h,之后使温度重新平衡至室温,平行配制两份(供试品溶液浓度:20 mg/mL)。
6)回收率测定溶液 称取400 mg供试品置于10 mL容量瓶中,分别用浓度水平为30%、80%、100%和120%的系列对照品储备溶液溶解并定容至刻度,混匀,再准确移取该溶液500 µL和衍生剂500 µL到1.5 mL液相小瓶,混匀,并将混合物加热至100 ℃持续1 h,之后使温度重新平衡至室温,平行配制三份。
2 结果
2.1 专属性
开发的LCMS方法的专属性是通过噁拉戈利钠和对照品溶液的保留时间确定的,采用常规反相色谱体系在C18色谱柱上进行分离,硼酸酯的保留时间约为3.6 min(图2)。低比例有机相(8%乙腈)主要用于洗脱硼酸酯,高比例有机相(95%乙腈)用于洗脱噁拉戈利钠,噁拉戈利钠的保留时间约为10.4 min和10.6 min(注:出现双峰是因为噁拉戈利钠化学结构中含有刚性环的手性N),硼酸酯和噁拉戈利钠都能达到基线分离,且空白溶液对主峰的检测无干扰,表明本方法专属性良好。注意事项:由于噁拉戈利钠浓度很高,为避免污染质谱系统,建议将流路设置为“引入废液”。
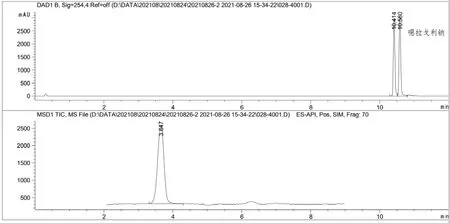
图2 化合物HPLC图
2.2 标准曲线与灵敏度
本方法的线性研究是通过从LOQ水平到150%水平浓度的六种杂质浓度溶液进行的。按照上述对照品溶液的制备过程和仪器方法参数进行LCMS分析,对照品溶液由低浓度到高浓度进样,在24.3~121.6 ng/mL的浓度范围内,通过最小二乘回归分析寻找“峰面积”和“杂质浓度”之间的关系,回归方程:y=430.447 5x+ 2 244.665 7,相关系数r为0.998 4(n=6)。表明在指定的浓度范围内两个参数之间线性关系良好。
本方法LOQ可达24.3 ng/mL(信噪比大于10)。将对照品溶液进一步稀释至信噪比约3时所得的浓度为本方法的LOD 8.1 ng/mL(信噪比大于3),满足法规规定的限量标准0.000 38%。
2.3 重复性
100%浓度水平(81.05 ng/mL)的对照品溶液六次重复进样,2-氟-3-甲氧基苯硼酸的保留时间RSD为1.0%和峰面积RSD为1.1%,在可接受的范围内,满足方法要求。
2.4 准确度
使用一式三份的回收率溶液评估30%、80%、100%和120%浓度水平的2-氟-3-甲氧基苯硼酸的准确性。计算得到其回收率范围为90.4%~106.5%。表明该方法回收率较好。
2.5 样品测试
选取公司制备的不同批次噁拉戈利钠原料药和粗品,制备成供试品溶液后进行LCMS分析测试,样品中均未检出2-氟-3-甲氧基苯硼酸。
2.6 溶液稳定性
存放于LC样品瓶中的对照品和供试品溶液于室温下加盖密封放置不同时间时进行测定,结果没有显着变化(表1),表明溶液于室温放置16 h稳定。

表1 溶液放置不同时间对2-氟-3-甲氧基苯硼酸测定的影响
2.7 衍生化试剂的选择
本研究选择N-甲基亚氨基二乙酸作为衍生化试剂是因为它方便易得,通用性好,与2-氟-3-甲氧基苯硼酸反应只生成一种衍生物,且能迅速被定量。为了使微量基因毒性杂质的转化完全,衍生化试剂与微量杂质摩尔比在几万到百万倍不等,本研究表明5 mg/mL衍生化试剂为HPLC上分离的合适浓度范围,浓度太高容易导致色谱柱过载,影响色谱柱性能。
3 讨论
我们开发了一个简单的衍生化方法即可测定原料药或原料药中间体中低至0.000 1%~0.000 5%的非含氮芳香族硼酸的测定方法。基于对2-氟-3-甲氧基苯硼酸化学结构和分子特性的理解,利用化学衍生化和铵离子螯合与LCMS相结合可以建立高灵敏的分析方法,该方法也可适用于其他药物中硼酸类基因毒性杂质的分析。本研究采用配备常规ESI源的LCMS单四级杆分析系统,用铵离子螯合和化学衍生法检测噁拉戈利钠中的基因毒性杂质2-氟-3-甲氧基苯硼酸含量,灵敏度高、方法简单、结果可靠、重现性好,满足目前法规规定的限量要求。另外,通过将仪器设置为将高浓度药品基质引入废液,可减少对质谱系统的污染。因此,该方法非常适合对噁拉戈利钠中潜在的痕量基因毒性杂质2-氟-3-甲氧基苯硼酸进行日常定量分析。
猜你喜欢
现代畜牧科技(2021年11期)2021-12-21 06:11:08
天津医科大学学报(2021年2期)2021-03-29 05:31:16
设备管理与维修(2019年9期)2019-09-12 07:44:02
云南医药(2019年3期)2019-07-25 07:25:14
中国畜牧兽医文摘(2015年9期)2015-12-29 03:38:09
大连工业大学学报(2015年4期)2015-12-11 04:06:50
中国塑料(2015年7期)2015-10-14 01:02:48
肿瘤预防与治疗(2015年1期)2015-09-26 07:26:19
应用化工(2014年12期)2014-08-16 13:10:46
湖南农业科学(2014年7期)2014-02-27 14:31:48
