
气相色谱法测定蔬菜中农药残留量的测量不确定度评定
2022-08-23 09:23:44张丽芳杜瑞焕郑百芹
计量学报 2022年7期
张丽芳, 张 亮, 杜瑞焕, 郑百芹, 刘 洋
(1.唐山市食品药品综合检验检测中心,河北 唐山 063000;2. 河北省农产品质量安全检测技术创新中心,河北 唐山 063000; 3.唐山市功能性农产品产业技术研究院; 河北 唐山 063000)
1 引 言
有机氯农药化学性质稳定,降解缓慢,易溶解于脂肪,可造成生物体的积累,对环境污染严重,主要有六六六、滴滴涕、腐霉利等。拟除虫菊酯类农药为人工合成的一类杀虫剂,毒性低,易分解,无蓄积,应用广泛,主要有联苯菊酯、氯菊酯、溴氰菊酯等[1]。农产品中过量的农药残留会对人体产生急性或慢性的毒害[2],因此农药残留问题越来越受关注,对检验结果的准确性要求也越来越高[3~5]。实验过程中存在多种不确定因素,通过不确定度来源的分析和量化评定,可提高数据的精准度。尤其当检测值与限量值接近时,不确定度可作为合格判定的重要参考依据,将最大限度地减少最大残留限量附近残留水平合规决定中可能存在的争议[6~8]。
蔬菜中农药残留量的测定多采用我国农业行业标准NY/T 761—2008《蔬菜和水果中有机磷、有机氯、拟除虫菊酯和氨基甲酸酯类农药多残留测定》中的方法,很多学者针对该方法进行了测量不确定度的评定。殷朝珍[9]等评定黄瓜中的甲胺磷、毒死蜱、地虫硫磷残留量检测的扩展不确定度均为0.002 mg/kg;洪泽淳[10]等评定蔬菜中16种有机磷农药残留的扩展不确定度为0.013 0~0.051 8 mg/kg;王璐[11]等评定蔬菜中甲胺磷、甲氰菊酯和甲萘威残留量检测的相对扩展不确定度分别为5.70%、5.66%和6.30%;柴宗龙[12]等评定黄瓜中丙溴磷农药残留量的扩展标准不确定度0.04 mg/kg;贺玲[13]等评定白菜中毒死蜱残留量的扩展不确定度为0.003 5 mg/kg;罗彦平[14]等评定番茄中联苯菊酯残留量的扩展不确定度为0.006 4 mg/kg。大部分已有文献报道的评定方法存在一定的局限,如:忽略了试剂的膨胀,容量瓶等量器在计算不确定度过程中选择分布方式不明确,方法回收率没有作为评定的考虑因素等。并且气相色谱法不确定度的评定多局限于有机磷类,或者是单个农药的残留,同时评定有机氯、拟除虫菊酯类的研究较少。日常检测中,有机氯和拟除虫菊酯类多放在同一组中测定,研究多组分同时测定的不确定度更具有实用价值。
本实验以NY/T 761—2008为检测标准,采用气相色谱法对白菜中六六六、腐霉利、联苯菊酯、高效氯氟氰菊酯、氯菊酯、氯氰菊酯6种农药残留量的不确定度进行评定,确定影响检测结果的主要因素,为气相色谱法测定有机氯和拟除虫菊酯类农药残留量的测量不确定度评定提供参考。
2 材料与方法
2.1 材料与仪器
标准物质:α-六六六(批号:GSB05-2276)、β-六六六(批号:GSB05-2277)、γ-六六六(批号:GSB05-2278)、δ-六六六(批号:GSB05-2279)、腐霉利(批号:GSB05-2338)、联苯菊酯(批号:GSB05-2333)、高效氯氟氰菊酯(批号:GSB05-2305)、氯菊酯(批号:GSB05-2309)、氯氰菊酯(批号:GSB05-2308),购自农业农村部环境质量监督检验测试中心(天津),质量浓度为1 000 μg/mL,不确定度为 7 mg/L(k=2)。色谱纯乙腈、正己烷购自德国Merck。
Agilent 7820A型气相色谱仪(安捷伦科技有限公司,配ECD检测器);TTL-DC氮吹仪(北京同泰联科技发展有限公司);HR7682均质机(飞利浦公司)。
2.2 试验方法
2.2.1 样品处理方法
样品处理方法依据NY/T 761—2008第1部分中的第2种方法。
2.2.2 标准溶液的配制
分别吸取有机氯、拟除虫菊酯标准品溶液 0.5 ml,置于50 mL容量瓶中,用正己烷定容,配制成10 μg/mL的标准储备液。吸取标准储备液 0.5 mL 置于50 mL容量瓶中,用正己烷定容,制备成0.1 μg/mL的标准溶液。
2.2.3 色谱条件
色谱柱:HP-5毛细管柱(30 m×0.25 mm,0.25 μm);进样口温度:220 ℃;检测器温度:290 ℃;色谱柱流量:1 mL/min;色谱柱温度:初始温度 100 ℃,保持2 min,以6 ℃/min的速率升至270 ℃,保持8 min;分流比为10:1。
2.2.4 数学模型
试样中农药残留量X(mg/kg)的计算公式为
(1)
式中:Cs为标准浓度,μg/mL;Ai为样品溶液峰面积;As为标准溶液峰面积;V1为提取溶剂总体积,mL;V2提取溶液分取体积,mL;V3为定容体积,mL;m为试样质量,g;fR为回收率。
3 结果与分析
3.1 分析不确定度来源
对实验过程每一步的影响因素进行分析,结合式(1),可得不确定度的来源包括:标准物质母液、标准溶液配制、试样称量、样品前处理、仪器稳定性和回收率,详见图1。

图1 不确定度来源分析图
测量不确定度评定分为A、B两类[15]。A类评定:利用统计方法对多次测量的结果进行计算得到的不确定度,评定方法有贝塞尔公式法和极差法两种[16]。陈凌峰[17,18]认为使用贝塞尔公式估计的总体方差总是无偏的,不会给测量结果的合成标准不确定度带来原理性误差;而极差法估计的总体方差偏大,导致最终测量结果的合成标准不确定度偏大。本文A类评定选择贝塞尔公式法。B类评定:非统计方法得到的不确定度。标准溶液重复测定、样品溶液重复测定、回收率测定属于A类评定;标准品、标准溶液的配制、试样称量等属于B类评定。
3.2 标准溶液配制引入的不确定度
3.2.1 标准物质母液引入的不确定度

3.2.2 标准储备液配制引入的不确定度
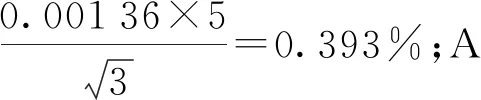
3.2.3 标准工作液配制引入的不确定度
配制标准工作液时,用1.0 mL的A级刻度吸管移取0.5 mL的10 μg/mL的标准储备溶液于50 mL容量瓶中,用正己烷定容,配制成0.1 μg/mL的储备液。该过程不确定度的引入涉及到试剂膨胀的影响、刻度吸管和容量瓶。引入的不确定度计算过程与标准储备液相同,urel(C3)=0.51%。
3.2.4 标准溶液配制的合成不确定度

3.3 样品称量引入的不确定度

3.4 样品的前处理引入的不确定度
3.4.1 提取液量取(V1)
提取液量取:用50 mL量筒量取40 mL乙腈试剂。乙腈在(20±5)℃条件下的体积膨胀系数为0.001 37/℃,引入的不确定度为0.395%。50 mL量筒满刻度最大误差为±0.50 mL,引入的不确定度分别为0.408%。提取液量取引入的合成相对不确定度为:urel(V1)=0.57%。
3.4.2 提取液分取(V2)
提取液分取:用10 mL刻度吸管吸取10 mL乙腈提取液。乙腈试剂膨胀引入的不确定度为0.395%,A级10 mL刻度吸管最大误差为±0.05 mL,引入的不确定度为0.204%。提取液分取引入的合成相对不确定度为:urel(V2)=0.44%。
3.4.3 浓缩定容(V3)
浓缩定容:将净化液氮吹浓缩至5 mL以下,然后用正己烷定容至5 mL。正己烷在(20±5)℃条件下的体积膨胀系数为0.001 36/℃,其相对不确定度为0.393%,A级5 mL刻度管最大误差为±0.025 mL,引入的不确定度为0.204%。浓缩定容引入的合成相对不确定度为:urel(V3)=0.44%。
3.4.4 前处理过程的合成不确定度
综合以上3个分量的不确定度,前处理过程引入的合成相对不确定度:
3.5 仪器稳定性引入的不确定度
气相色谱仪(GC)的稳定性主要体现在峰面积的差异。对6种有机氯、拟除虫菊酯类标准溶液和试样重复测试10次,用峰面积的相对标准偏差来表示GC测定引入的不确定度。按贝塞尔法计算。
(2)
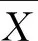
3.5.1 标准溶液峰面积引入的不确定度urel(As)
在重复性条件下,标准溶液连续重复进样10次,α-六六六、β-六六六、γ-六六六、δ-六六六、腐霉利、联苯菊酯、高效氯氟氰菊酯、氯菊酯、氯氰菊酯标准溶液重复测定引入的相对不确定度分别为:0.79%,1.37%,1.08%,0.84%,0.81%,0.71%,1.10%,1.27%和0.98%,详见表1。
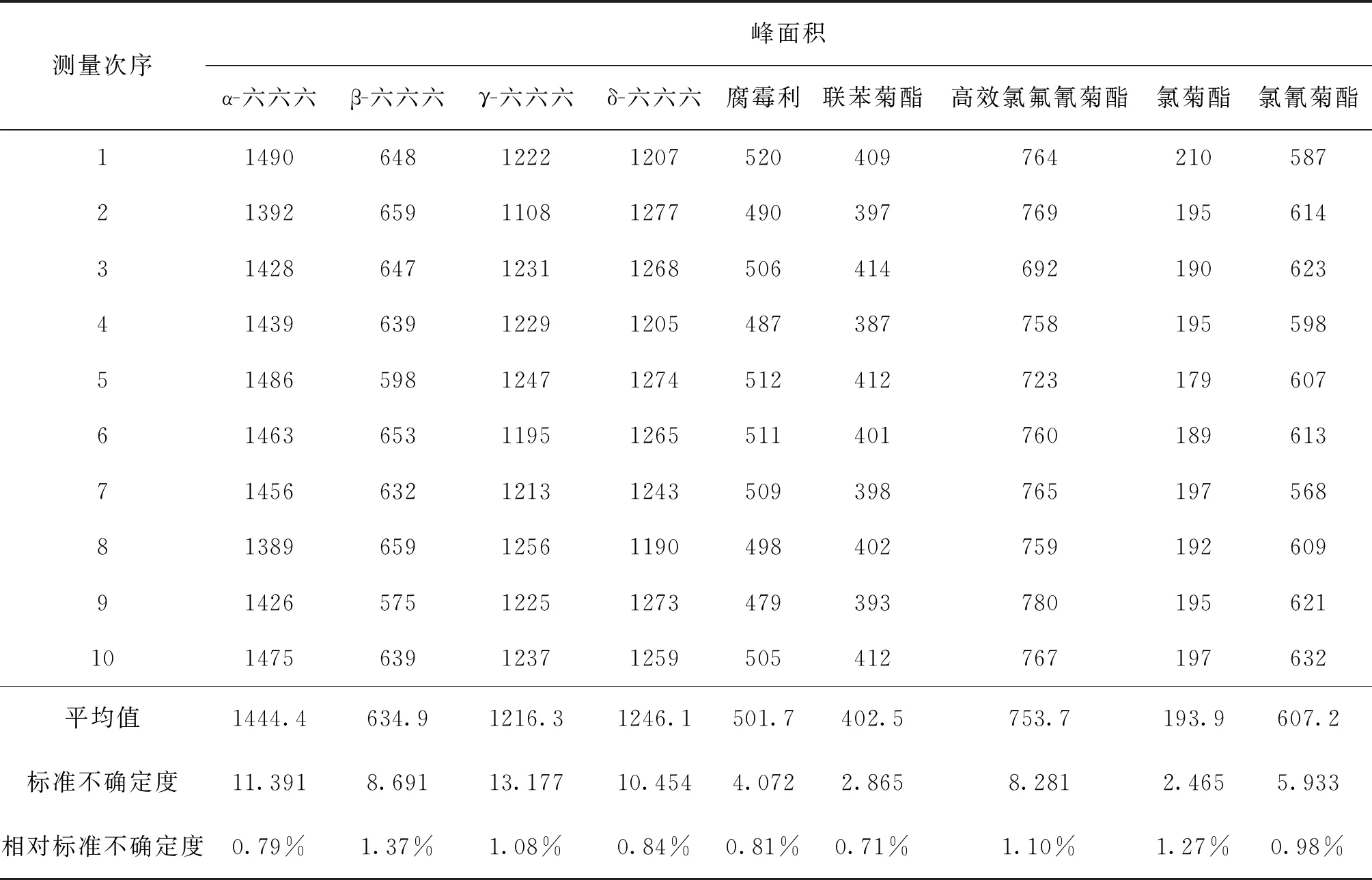
表1 标准溶液重复测定引入的不确定度
3.5.2 样品溶液峰面积引入的不确定度urel(Ai)
在重复性条件下,一份添加量为0.08 mg/kg的白菜样品,经处理后,连续重复进样10次,α-六六六、β-六六六、γ-六六六、δ-六六六、腐霉利、联苯菊酯、高效氯氟氰菊酯、氯菊酯、氯氰菊酯样品溶液重复测定引入的相对标准不确定度分别为:0.96%,1.15%,0.73%,0.79%,1.08%,0.92%,1.31%,1.77%和1.36%,详见表2。

表2 样品溶液重复测定引入的不确定度
3.6 回收率引入的不确定度urel(fR)
在0.08 mg/kg添加水平下测定6次,α-六六六、β-六六六、γ-六六六、δ-六六六、腐霉利、联苯菊酯、高效氯氟氰菊酯、氯菊酯、氯氰菊酯检测,回收率引入的相对标准不确定度分别为:2.01%,2.13%,1.72%,2.14%,1.51%,0.81%,1.23%,2.25%和1.97%,详见表3。
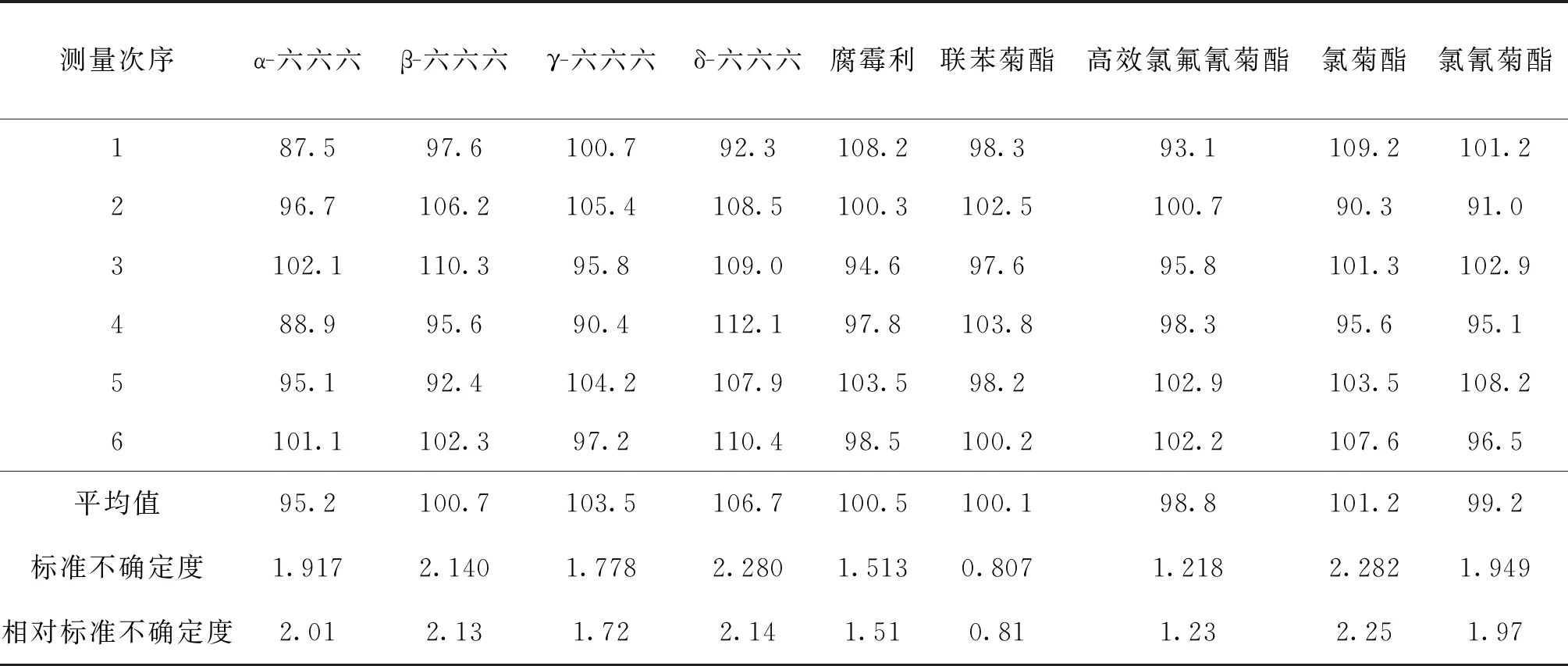
表3 回收率测定结果与不确定度
3.7 合成的不确定度
合成相对不确定度urel(X)计算公式如下:
(3)
计算结果见表4。影响合成不确定度的主要因素有:回收率测定、样品溶液的峰面积和标准溶液峰面积。回收率对α-六六六、β-六六六、γ-六六六、δ-六六六、腐霉利、氯氰菊酯残留量合成不确定度的贡献率分别为58%、50%、49%、63%、42%和48%,回收率在六六六、腐霉利和氯氰菊酯不确定度评定中占主要因素;样品溶液峰面积和标准溶液峰面积重复测定对联苯菊酯、高效氯氟氰菊酯和氯菊酯残留量测量不确定度的贡献率之和分别为40%、50%、42%,仪器的稳定性在这3种农药残留的不确定度评定中占主要因素。
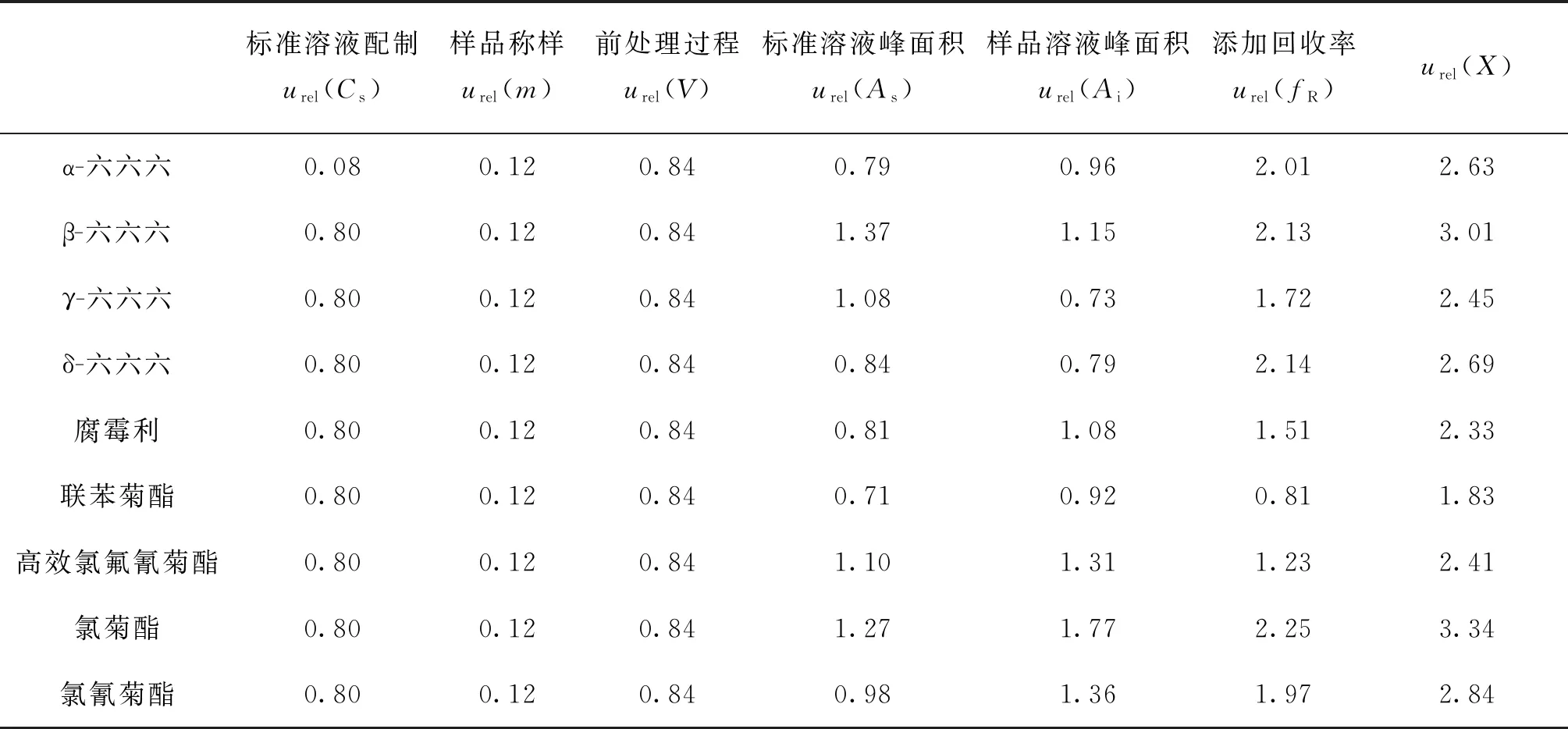
表4 各种农药残留量测定的相对合成不确定度
回收率体现了检测过程中对目标物的提取、净化效率,样品溶液峰面积、标准溶液峰面积主要体现仪器的稳定性,以上因素占合成不确定度的主要内容。黄荣浪[21]等认为不论试验结果是否折算回收率,回收率对试验结果的不确定度均存在显著影响;汪志威[22]等的研究结果表明对合成不确定度贡献最大的是样品溶液峰面积和回收率;以上结论均与本试验相同。文献[23,24]的研究认为,标准品配制、样品的称量与定容对不确定度贡献最大,分析原因,其标准溶液配制经过6次稀释,增加了标准品配制过程的不确定度;且测量氯氰菊酯和溴氰菊酯的相对标准不确定度较小,仅为1.86%和1.91%。本试验结果也体现出对合成不确定度较小的目标物,标准溶液的配制、前处理过程的定容和量取对合成不确定度的影响较大,如联苯菊酯的前处理过程的贡献率为21%。洪泽淳[10]等研究结果显示标准曲线的拟合对不确定度影响最大,但标准曲线不确定度计算模型未考虑待测样品实际含量的不确定性带来的影响。
本试验采用单点标准浓度校准的计算模型,降低标准曲线拟合带来的影响。称量过程对合成不确定的影响较小。各因素的不确定度贡献率统计见表5。
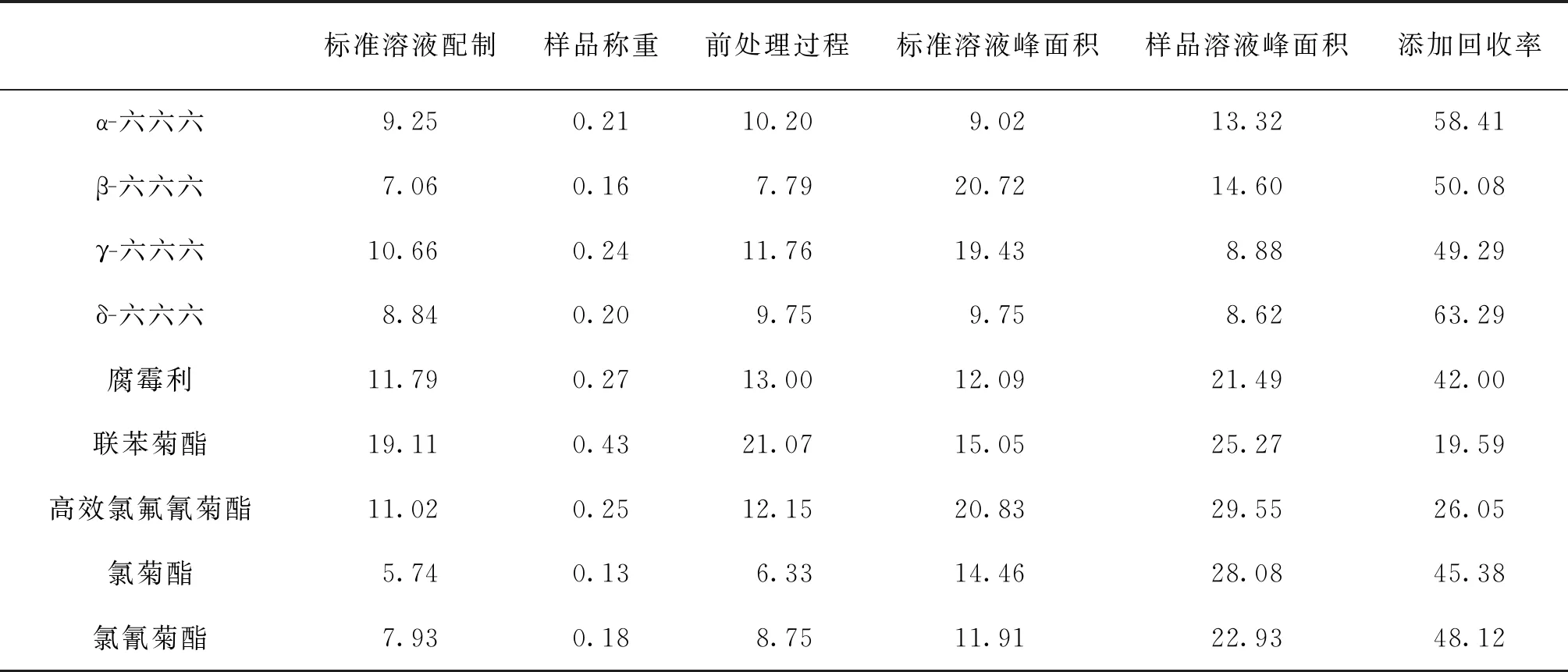
表5 各因素对合成不确定度的贡献率
3.8 扩展不确定度和检测结果
在95%置信概率下,取包含因子k=2,则在0.08 mg/kg添加水平下,白菜中各种农药残留量测定的检测值和扩展不确定度如表6所示。

表6 各种农药残留量测定的检测结果
4 结 论
测量不确定度的评定对检验结果的准确性可提供可靠的判定依据。本次试验结果显示影响上述6种农药残留量检测不确定度的主要因素为回收率和仪器稳定性。回收率的影响与基质的复杂性、基质效应、农药本身的化学性质等有关,可通过优化前处理方法降低回收率对测量不确定度的影响。仪器稳定性是影响不确定度的另一个主要因素。实验室应及时做好仪器的检定、期间核查和维护保养工作,使仪器维持在一个比较好的工作状态。对于标准品纯度、标准溶液的配制和前处理过程中的量取、定容等因素对不确定度的影响亦不能忽视。在日常检测工程中,尽可能减少稀释过程,所用精密量器应定期校准。测定不确定度应融入到整个检测过程中,合理运用,提高检测结果的准确性。
猜你喜欢
世界农药(2019年2期)2019-07-13 05:55:12
广东茶业(2019年2期)2019-06-18 10:24:24
农药科学与管理(2019年12期)2019-05-20 09:33:26
食品与机械(2019年1期)2019-03-30 01:14:36
中成药(2018年1期)2018-02-02 07:20:31
电源技术(2015年7期)2015-08-22 08:48:52
电测与仪表(2015年7期)2015-04-09 11:40:30
中国茶叶加工(2015年3期)2015-02-27 07:55:29
中国药业(2014年24期)2014-05-26 09:00:16
电测与仪表(2014年9期)2014-04-15 00:27:16
