
基于数据挖掘的二氢查尔酮衍生物降血糖构效关系研究
2022-08-23 13:32吕蔚
上海化工 2022年4期
吕蔚
上海市塑料研究所有限公司(上海 201700)
目前新药发现的流程为:药物作用靶点确定—先导化合物确定—构效关系研究与活性化合物筛选—候选药物选定。其中构效关系研究与活性化合物筛选往往需要投入巨额的资金且耗时往往数十年。为加快研究进展,国际医药巨头纷纷引入了虚拟药物筛选技术。虚拟药物筛选是在计算机上模拟药物构效关系,对化合物可能的生理活性作出预测,并据此对系列化合物进行有针对性的试验筛选,可以有效地缩短药物开发时间、降低开发成本。
糖尿病作为一种高发疾病,主要由于患者体内胰岛素缺乏或拮抗胰岛素的激素增加,或胰岛素在靶细胞内不能正常发挥生理作用,从而引起机体的葡萄糖、蛋白质及脂质代谢紊乱。二氢查尔酮类化合物被发现具有较好的胰岛素促泌作用,是一种很有发展前景的降血糖药物前驱体。对不同取代基的二氢查尔酮系列化合物开展虚拟药物筛选,对于开发活性功能更强、疗效更显著的糖尿病药物[1-8]具有重要意义。
本研究基于数据挖掘方法,通过对系列二氢查尔酮类[9-11]化合物的促胰岛素分泌和降血糖功效数据进行筛选,建立构效关系的预报模型,以达到提高药物开发效率的目的。
1 二氢查尔酮类化合物的胰岛素促泌功能
二氢查尔酮类化合物的基本结构如图1所示,其A,B环上的羟基与甲氧基的位置、个数、分子的对称性、空间位阻、内氢键等参数直接影响化合物促胰岛素分泌的强弱。
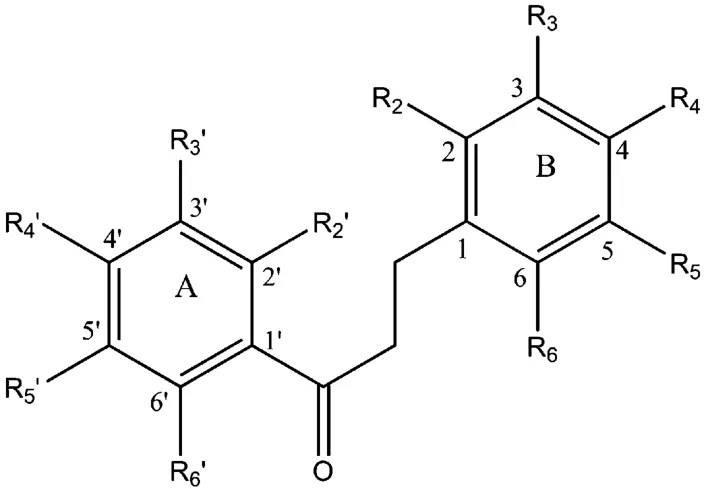
图1 二氢查尔酮类化合物的基本结构
在本研究中,以前期合成的38个二氢查尔酮类化合物的物理化学参数作为分子描述符,对照其胰岛细胞胰岛素促泌性能进行构效关系研究[12-13]。采用数据挖掘中基于最大相关性和最小冗余性的CFS(基于相关性的特征选择算法)[14-15]进行变量的筛选;用SVM(支持向量机)回归算法[16-18]进行建模,并用留一法验证模型的性能。
2 软件和方法
2.1 计算软件及算法
采用ChemOffice7.5软件来绘制38个二氢查尔酮类化合物的分子结构,使用MODEL软件计算其物理化学分子描述符作为变量,选用Weka软件进行变量筛选,最后利用上海大学在线数据挖掘平台OCPMDM的SVM回归功能,建立并优化二氢查尔酮衍生物降血糖构效关系模型。
2.2 数据集
将38个二氢查尔酮类化合物随机分为A,B两组,A组19个化合物的胰岛素分泌量为训练集样本,见表1;B组19个化合物为测试集样本,见表2。化合物的活性胰岛细胞胰岛素分泌量数据由上海大学生命学院提供。

表1 A组19个二氢查尔酮化合物名称、结构式及胰岛素分泌量训练集样本
2.3 分子描述符的筛选
本研究采用新加坡国立大学的MODEL软件,计算了表1和表2中所有分子的3大类45种分子描述符。所计算的分子描述符主要为电负性参数、立体参数和热力学参数,具体见表3。
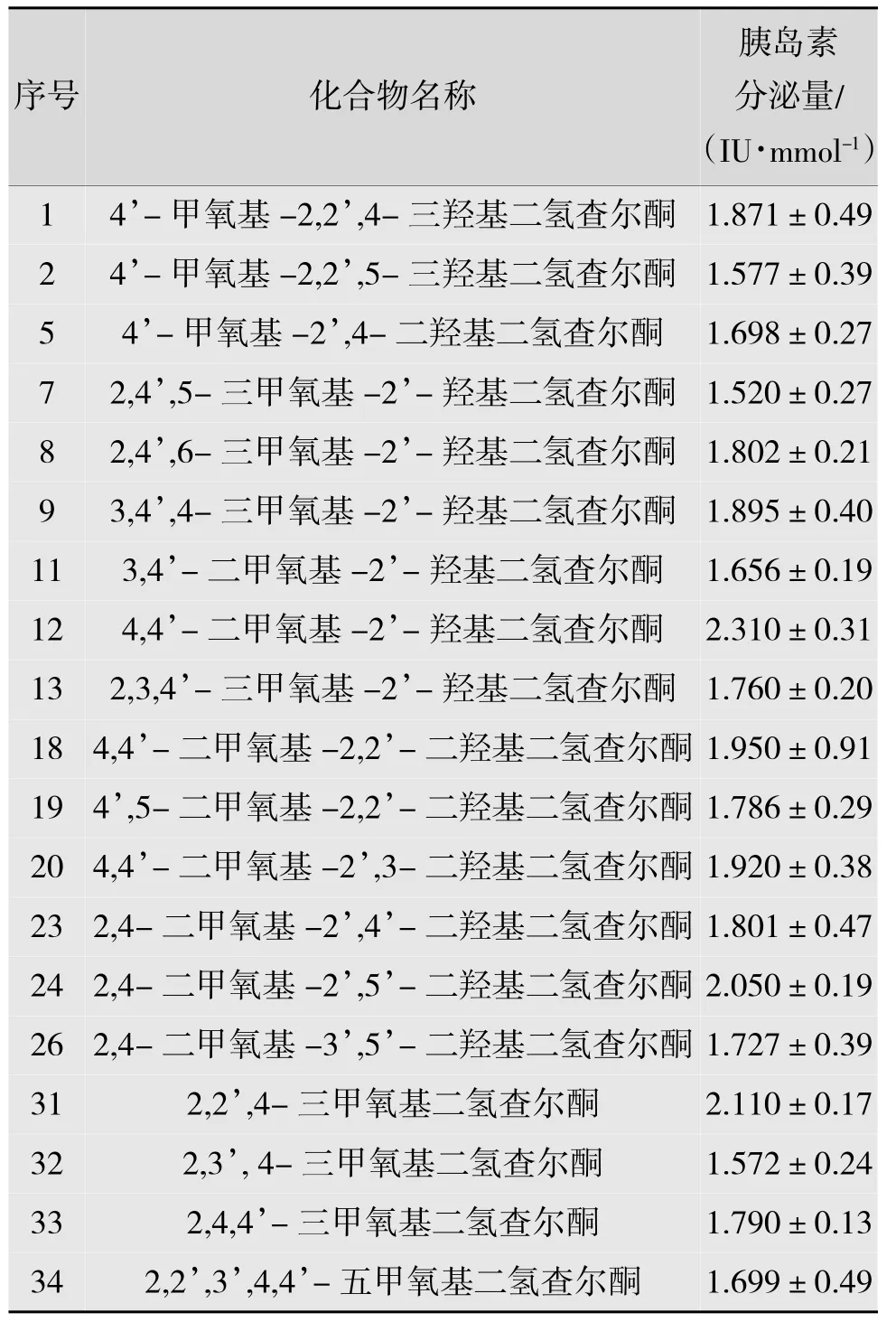
表2 B组19个二氢查尔酮化合物的名称、结构式及胰岛素分泌量测试集样本

表3 分子描述符的类别与名称
随后基于CFS算法进行了上述分子描述符的筛选,挑选出了4个特征参数,它们分别为电负性参数中的电子能量(Electronic energy)、立体参数中的疏水性(logP)、摩尔折射率(MR)以及热力学参数中的分子直径(Diameter),并以此为基础建立SVR预测模型。
2.4 SVR模型的建立
为获得最佳的模型泛化能力,需要对SVR模型的函数进行选择、参数进行优化。
图2至图4分别给出了线性核函数、多项式核函数以及径向基核函数的优化过程随着参数容忍因子C(C=1~100,step=1)和不敏感损失函数ε(ε=0.005~0.500,step=0.005)采 用 留 一 法 交 叉 验 证(LOOCV)时的均方根误差(RMSE)变化情况。从图可以看出,3个函数中,径向基核函数的误差最小、验证准确率最高,所以本研究选择径向基核函数开展建模。
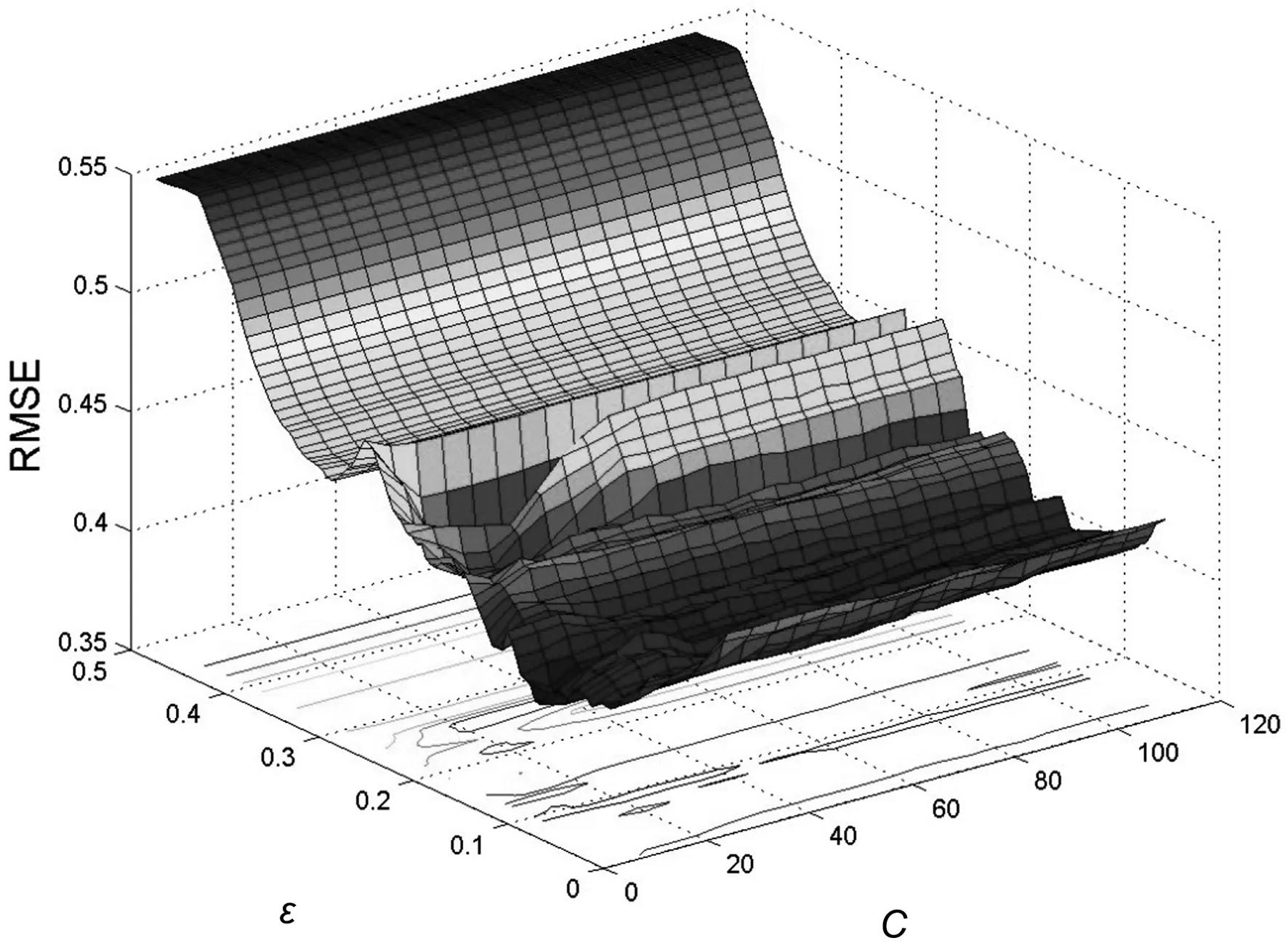
图2 线性核函数随C(C=1~110,step=1)和ε(ε=0.005~0.500,step=0.005)LOOCV的RMSE变化情况

图3 多项式核函数随C(C=1~100,step=1)和ε(ε=0.005~0.500,step=0.005)LOOCV的RMSE变化情况

图4 径向基核函数随C(C=1~100,step=1)和ε(ε=0.005~0.500,step=0.005)LOOCV时的RMSE变化情况
在图5和图6中,分别研究了径向基核函数条件下,容忍因子C、不敏感损失函数ε与gamma参数g对模型预报性能的影响。从图5和图6可得:当C=8.2,ε=0.1和g=0.01时,RMSE取得最小值,所以选择该组参数进行建模。

图5 径向基核函数随C(C=5~110,step=5)和g(g=0.05~1.1,step=0.05)LOOCV的RMSE变化情况

图6 径向基核函数随g(g=0.1~1.1,step=0.1)和ε(ε=0.01~0.300,step=0.001)LOOCV的RMSE变化情况
2.5 模型的应用研究
通过变量筛选,挑选出电子能、疏水性、摩尔折射率以及分子直径,并以此建立了预测模型。由于所建立模型是非线性的,无法直接从模型中分析各变量对模型预报准确率的影响,但可以敏感性分析来实现对不同分子结构二氢查尔酮类化合物降血糖功效的预测,见图7至图10。
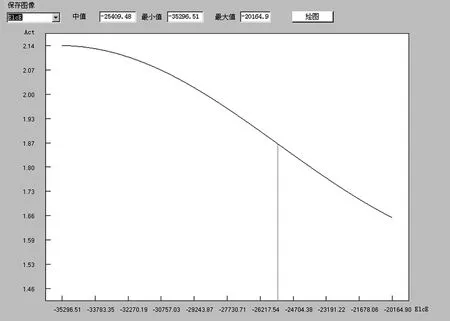
图7 二氢查尔酮化合物促胰岛素分泌活性与电子能量的关系

图10 二氢查尔酮化合物促胰岛素分泌活性与分子直径的关系
由图7可见,随着电子能的升高,化合物的活性值逐渐降低。因此,可以初步认为,不考虑其他因素,一些低电子能的二氢查尔酮类似物可能具有更高的活性。
疏水性logP反映了化合物在人或动物体内的富集和积累。由图8可见,随着疏水性的增加,化合物的活性逐渐升高,但在疏水性达到2.5时,出现了一个拐点,随后化合物的活性随着疏水性的增加而逐渐降低。因此,当疏水性为2.5左右时所设计的化合物可得到比较高的活性值。

图8 二氢查尔酮化合物促胰岛素分泌活性与疏水性的关系
在QSAR研究中,常用摩尔折射率来表征取代基的立体结构或者空间效应。由图9可见,随着摩尔折射率的增大,活性值逐渐上升;但当摩尔折射率为9.3时,出现了一个拐点,随后活性值随摩尔折射率的增大而平缓下降。因此,可以认为摩尔折射率9.3附近的化合物可能具有令人满意的活性。
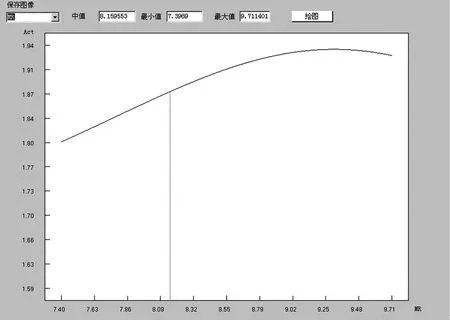
图9 二氢查尔酮化合物促胰岛素分泌活性与摩尔折射率的关系
由图10可见,随着分子直径的增大,活性值逐渐上升;当分子直径为13.7时,出现了一个拐点,随后活性值随分子直径的增大而平缓下降。可以认为分子直径13.7附近的区域内,所设计的化合物可能具有令人满意的活性。
使用敏感性分析法分析各变量对模型预报准确率的影响,首先以二氢查尔酮活性SVR模型的留一法交叉验证的平均相对误差作为多模型优劣的判据,通过留一法验证预报的准确率,得到A组19个化合物活性值预报的平均预测偏离度为10.67%,然后以B组19个二氢查尔酮化合物作为独立测试集,得到平均预测偏离度为11.63%(见表4)。
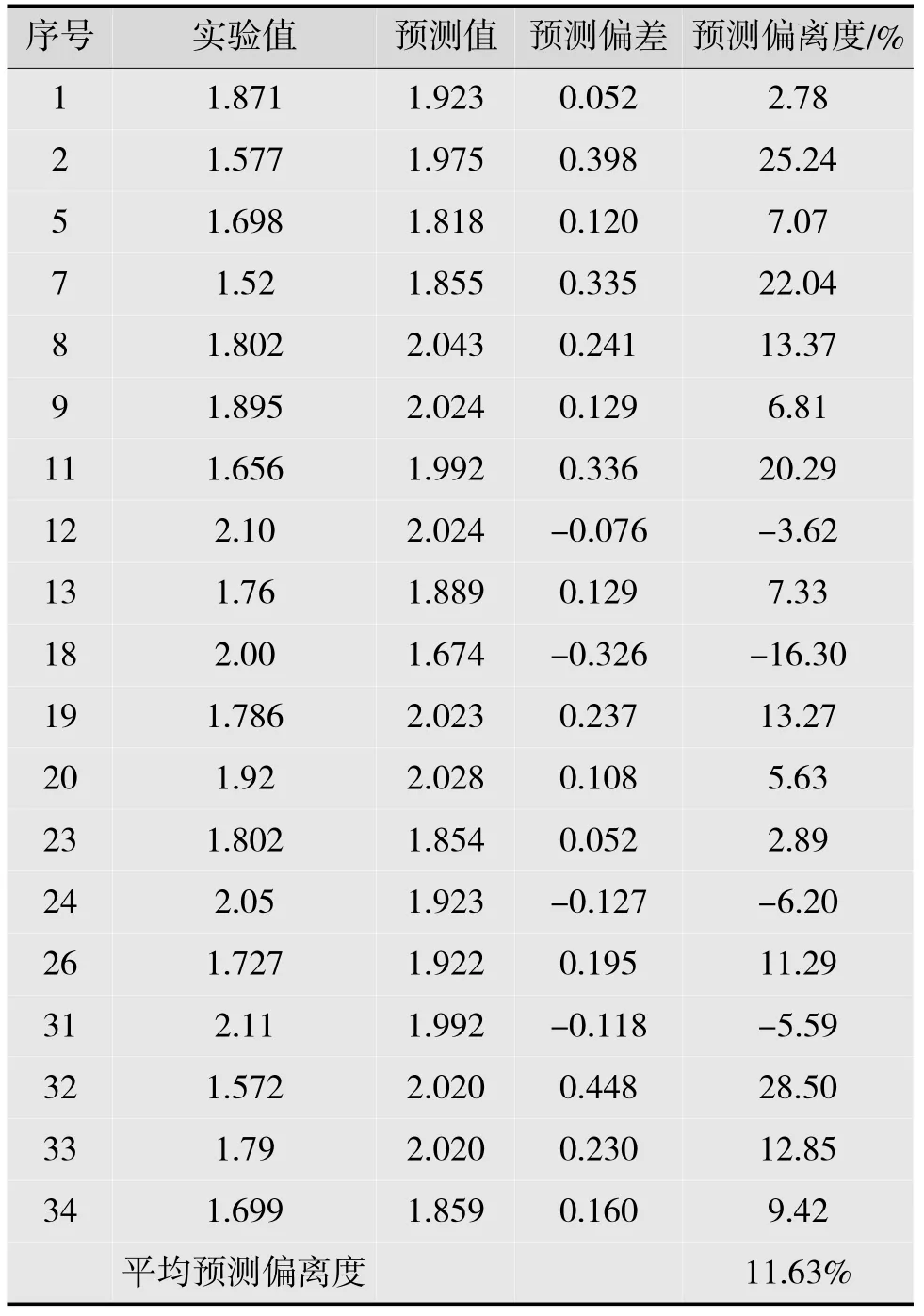
表4 B组19个二氢查尔酮化合物测试集预报率的预测偏离度
A,B组的平均预测偏离度相近,表明所建立的预测模型可以得到相对满意的预报准确率。
3 总结
本研究通过数据挖掘,采用SVR算法研究二氢查尔酮衍生物的构效关系,建立了该类化合物降血糖性能与其分子参数之间的模型,为进一步筛选出生物活性强的化合物提供了依据,利用该模型将有望筛选出降血糖更强的二氢查尔酮化合物。
猜你喜欢
当代化工研究(2022年19期)2022-11-04
中学生数理化·中考版(2021年12期)2021-12-31
农业与技术(2021年3期)2021-12-06
中学生数理化·中考版(2021年11期)2021-12-06
世界科学技术-中医药现代化(2021年7期)2021-11-04
农村青少年科学探究(2020年5期)2020-08-18
科学大观园(2019年23期)2019-09-10
新民周刊(2018年8期)2018-03-02
饮食科学(2017年12期)2018-01-02
中学化学(2017年6期)2017-10-16
