
HLB净化/高分辨液质联用法快速筛查动物肝脏中45种兽药残留
2022-08-17 05:56郭添荣吴文林万渝平彭海川郑漫江裴博文
中国食品学报 2022年7期
郭添荣,吴文林,万渝平,张 崟,彭海川,郑漫江,裴博文
(1 成都市食品检验研究院 成都 611130 2 成都大学肉类加工四川省重点实验室 成都 610106 3 中国科学院成都生物研究所 成都 610041 4 中国科学院大学 北京 100049 5 中国检验检疫科学研究院 北京 100176)
兽药在防治动物疾病和促进动物生长等方面发挥着重要作用[1],然而,由兽药残留引发的食品安全问题屡见报道。欧美国家早已颁布了相关禁限令[2],我国也制定了如农业部第235 号公告以及GB31650-2019 等一系列限量标准[3],明确规定限制或禁止兽药用于可食动物的养殖[4],然而,在利益驱使下非法滥用现象普遍存在[5-6]。研究表明,残留兽药超标对消费者健康、生态环境和经济贸易都有负面影响[7]。动物肝脏中含有丰富的蛋白质和维生素,具有补气血、健肝脾的作用[8],是人们日常饮食主要来源之一。动物肝脏样品基质十分复杂且内源成分结构各异,传统单一复杂的检测方法难以满足兽药多残留监管的需要,有必要建立一套动物肝脏中兽药多残留的快速筛查方法。
目前药物残留检测方法主要有免疫分析法[9-10]、高效液相色谱法[11-12]、液相色谱-串联质谱法[13-15]等。其中,免疫分析法受活性酶种类限制且假阳性高而逐渐被取代。高效液相色谱法仅能分离几种特定的物质,且抗干扰能力不高,应用受限。液相色谱-串联质谱法虽为主流检测方法,但受分析模式和分辨率限制,难以实现对复杂样品的高通量多目标分析[16-17]。超高效液相色谱-高分辨质谱联用法将液相色谱与高分辨质谱有机结合,面对复杂基质可实现无标准品对照情况下对化合物多目标快速筛查[18-19]。兽药痕量分析过程中的样品前处理方法主要有固相萃取、基质固相分散萃取以及加速溶剂萃取和QuEChERS 等[20]方法,其中基于通过式固相萃取技术,在保证良好净化效果的基础上,可免去活化与平衡等步骤,直接上机测试,适合于大规模样品检测的快速净化,现研究报道多应用于肉类食品与保健品中兽药残留的筛查研究[21-22],而用于动物肝脏中兽药残留的筛查还未见报道。
本文将固相萃取(PRiME HLB)净化与超高效液相色谱-四极杆/静电场轨道阱高分辨质谱(Ultra performance liquid chromatography -quadrupole-Orbitrapl high resolution mass spectrometry,UHPLC-Q-Orbitrap HRMS)结合,旨在筛查6 种大宗动物肝脏中45 种兽药残留,为大批量动物肝脏样品中兽药残留的快速筛查与确证提供技术参考。
1 材料与方法
1.1 材料、试剂及仪器与设备
动物肝脏54 批次(猪肝、牛肝、羊肝、鸡肝、鸭肝和鹅肝各9 批次),购自当地农贸市场和生鲜超市。
乙腈、甲醇(质谱级),美国Thermo Scientific公司;甲酸、乙酸铵(质谱级),美国Sigma Aldrich公司;陶瓷均质子(100/pk),美国Agilent 公司;Oasis®PRiME HLB 固相萃取柱,美国waters 公司;超纯水(电阻率为18.2 MΩ/cm,25 ℃),美国Millipore 公司;45 种兽药标准品(红霉素、吉他霉素、林可霉素、泰乐菌素、替米考星、泰万菌素、竹桃霉素、多拉菌素、伊维菌素、罗红霉素、金霉素、强力霉素、四环素、土霉素、差向土霉素、差向金霉素、去甲基金霉素、氨苄西林、苯唑西林、氯唑西林、双氯西林、青霉素、阿莫西林、氯丙嗪、异丙嗪、乙酰丙嗪、赛拉嗪、特拉唑嗪、哌唑嗪、头孢匹啉、头孢喹肟、头孢噻呋、头孢氨苄、头孢唑啉、氟虫腈、氟虫腈砜、氟虫腈亚砜、氟甲腈、苯乙醇胺A、金刚烷胺、金刚乙胺、氟苯尼考胺、可乐定、安普乐定、替扎尼定,纯度均≥95.0%),德国Dr.Ehrenstorfer GmbH 公司。
Vanquish UHPLC-Q Exactive Plus 高分辨质谱系统,美国Thermo Fisher 公司;浓缩氮吹仪,美国Biotage Caliper Zymark 公司;ME203 型电子天平,瑞士METTLER TOLEDO 公司;Milli-Q 超纯水系统,美国Millipore 公司;3K15 高速冷冻离心机,美国SigmaAldrich 公司;ORTEX3 漩涡混匀器、T18 basic 均质机,德国IKA 公司。
1.2 试验方法
1.2.1 样品前处理 准确称取2.00 g(精确至0.01 g)匀浆试样,置于50 mL 聚丙烯(PP)离心管中,加入80%乙腈水溶液(含0.2%甲酸)10 mL,静置15 min 后加入两粒陶瓷均质子,快速剧烈振摇并旋涡振荡30 min(2 000 r/min),使样品充分散开,9 500 r/min 高速离心5 min,取上清液待净化。取5 mL 准确量取的上清液过6 mL 200 mg 的Oasis®PRiME HLB 固相萃取柱,取2 mL 流出液在氮气下吹干至1 mL,初始流动相定容,过0.22 μm 有机滤膜后上机。
1.2.2 仪器条件 UHPLC 条件:Waters Acquity BEH C18色谱柱(2.1 mm×100 mm,1.7 μm)。柱温:25 ℃。流速:0.5 mL/min。流动相A 为0.1%甲酸-乙酸铵(20 mmol/L)水溶液,流动相B 为乙腈。进样量:10 μL。梯度洗脱程序见表1。
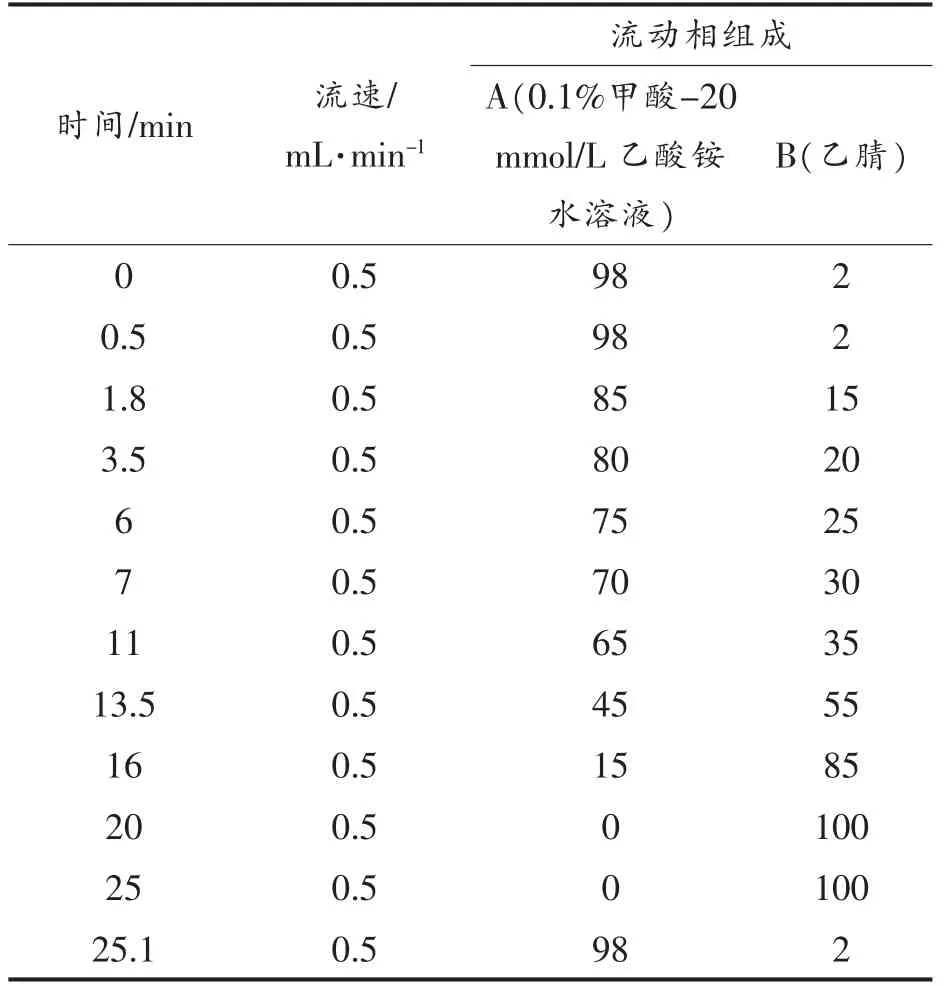
表1 液相色谱流动相梯度洗脱程序Table 1 Liquid chromatography mobile phase gradient elution procedure
HRMS 条件:加热电喷雾离子源;一级全扫描/数据依赖二级扫描模式(Full MS/dd-MS2);喷雾电压:3.5 kV(+),-3.0 kV(-);离子传输管和加热器温度:325 ℃和450 ℃;鞘气和辅助气(N2)流速:35 arb 和10 arb;采集范围:100~1 000 m/z;分辨率:75 000(Full MS)、17 500(dd-MS2);C-Trap 中离子数最大容量(AGC target):3×106(Full MS)、2×105(dd-MS2);C-Trap 中最大注入时间:100 ms(Full MS)、50 ms(dd-MS2)。
1.2.3 数据库的建立与应用 基于TraceFinder 4.1 和mzVault 软件搭建数据库。基于TraceFinder 4.1 软件,在收集化合物基本信息的基础上,利用仪器分析获取各化合物的色质谱信息 (见表2),得到可供快速筛查的Database。母离子经不同碰撞能量裂解后,基于mzVault 软件搭建HCD 碎片离子谱图匹配确证的谱图库。

表2 45 种兽药化合物的色质谱指纹信息表Table 2 Fingerprint information table of 45 veterinary drug compounds by color mass spectrometry
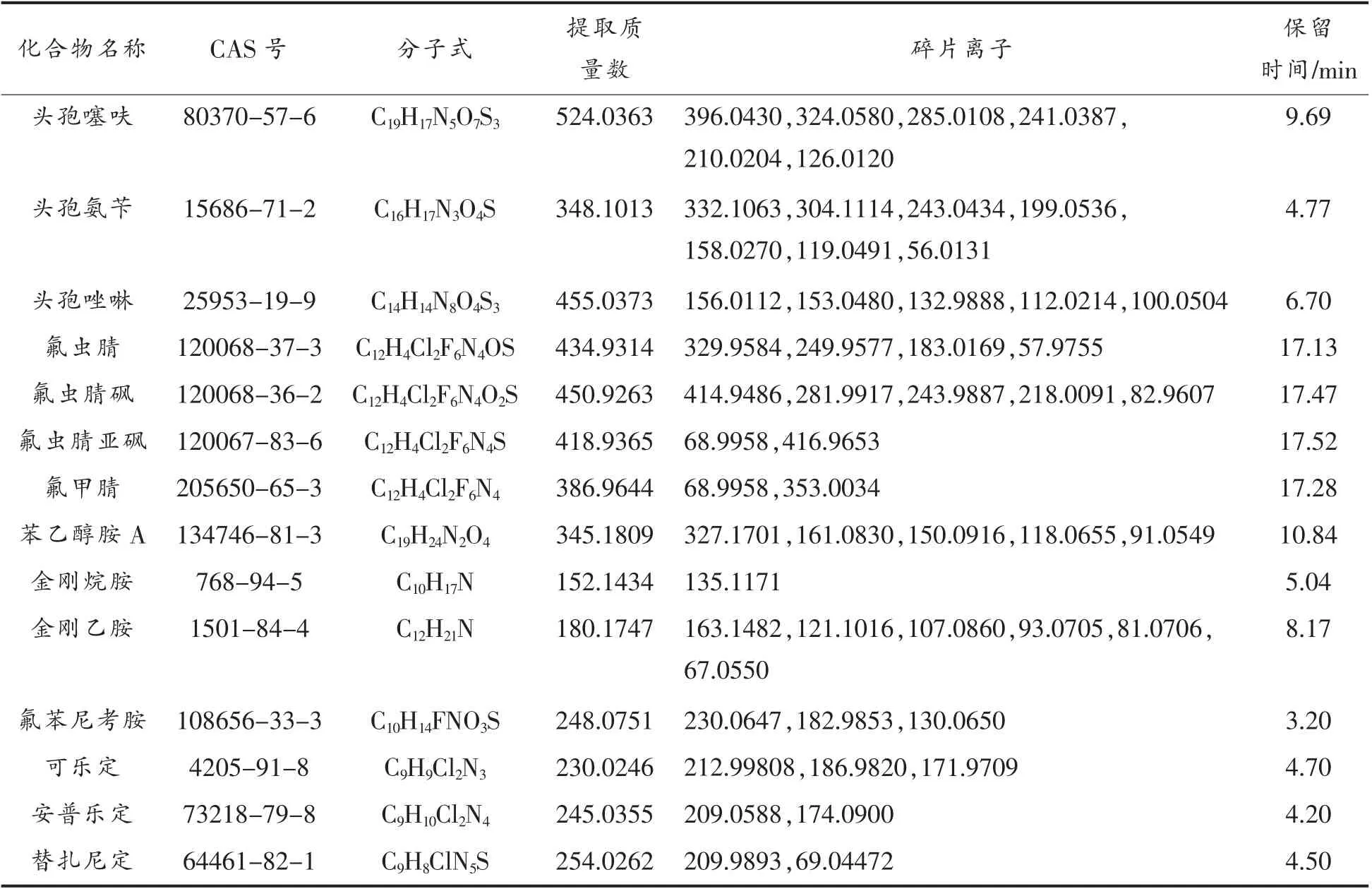
(续表2)
1.2.4 基质效应的计算 将45 种兽药化合物标准品制备为标准溶液,根据甲醇纯溶剂和基质溶液中同一物质的标准曲线斜率之比,对化合物的基质效应进行评价。基质效应(ME%)计算公式如下:

评价依据:1)80%≤ME%≤120%,基质效应可忽略;2)ME%<80%或ME%>120%,存在基质效应。
2 结果与分析
2.1 优化的前处理条件
2.1.1 提取溶剂及提取次数 考察了6 种常见提取剂对6 种动物肝脏基质中45 种兽药的提取效果,见图1a。乙酸乙酯和纯乙腈共提物较多且纯乙腈更容易导致样品结块;乙腈水溶液提取效率明显高于乙酸乙酯和纯乙腈,这可能与乙睛水溶液具有较好的蛋白沉淀作用有关;80%体积分数的乙腈水溶液提取效果率高于90%体积分数的,可能是由于高浓度乙腈下目标化合物被大量变性蛋白质包裹而不易被解离提取;而在80%体积分数的乙腈水溶液中添加0.2%甲酸的提取效率高于1.0%甲酸,可能是高浓度甲酸降低了样品溶解性,影响离子化效率。最终选择0.2%甲酸-80%乙腈水溶液为提取溶剂。
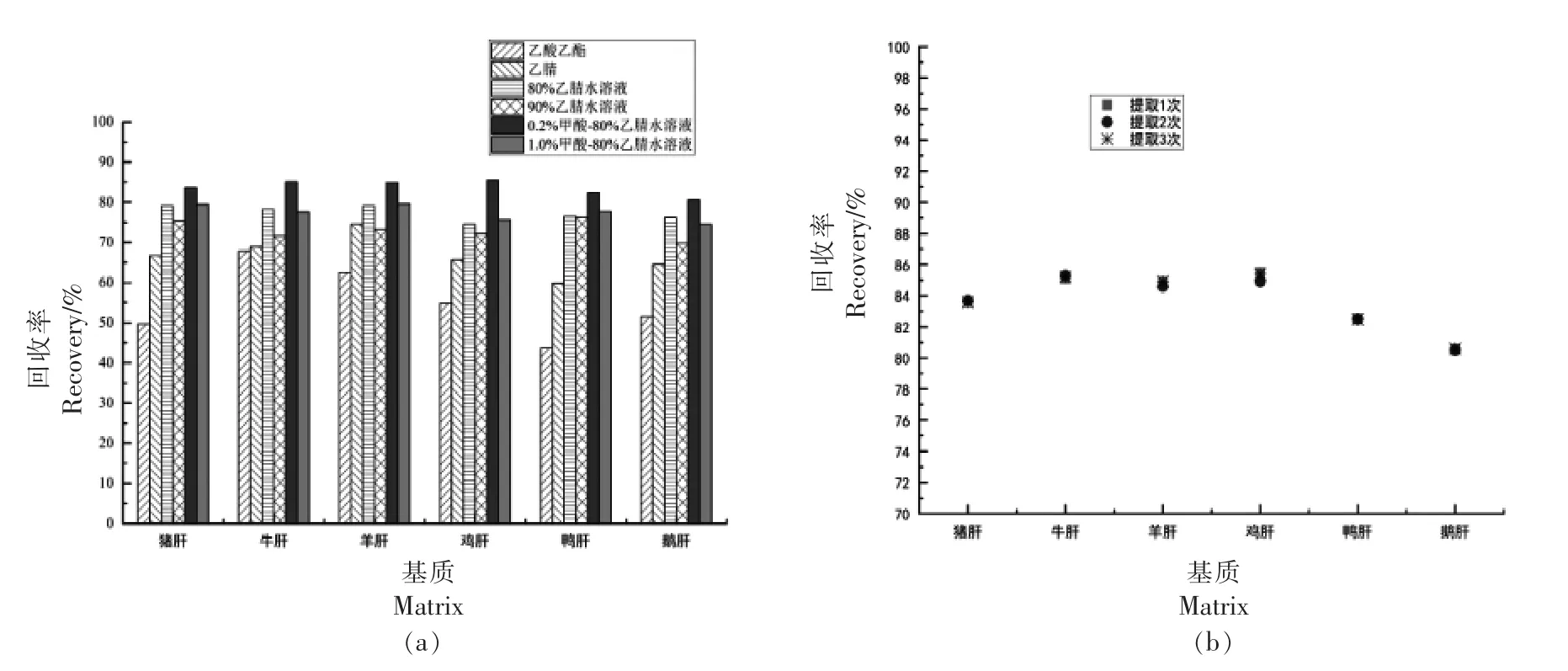
图1 不同提取溶剂(a)及提取次数(b)对45 种兽药回收率的影响Fig.1 Effects of different extraction solvents (a) and extraction times (b) on the recovery rates of 45 veterinary drugs
肝脏样品为固体基质,与提取溶液不混溶。在选取提取溶剂的基础上分3 次提取,以6 种肝脏基质中45 种兽药的平均回收率为提取效果评价依据,考察提取次数对兽药提取效果的影响。结果发现,在提取1 次、2 次和3 次情况下,样品中药物的平均回收率无显著性差异,可能是因为用0.2%甲酸-80%乙腈水溶液提取1 次已实现对药物的完全提取。从工作效率和成本等方面综合考虑,最终选择1 次提取。
2.1.2 固相萃取柱及填料量 动物肝脏基质十分复杂,样品经提取后仍存在杂质,需净化处理。分别考察C18、Oasis HLB、Oasis PRiME HLB 和Captiva EMR Lipid 4 种萃取柱对6 类基质中45种兽药的分离净化效果。由图2a 可知,采用C18时,6 类肝脏产品中45 种兽药的平均回收率最低,可能是由于传统C18吸附剂难以同时兼顾所有兽药性质,Oasis®PRIME HLB 的净化效果均明显优于Oasis®HLB 和Captiva EMR Lipid,表明Oasis®PRIME HLB 能够更好地吸附样品中的磷脂和蛋白质等杂质。PRIME HLB 为多兽残专用且适用于高分辨质谱的萃取柱,该柱可免去活化与平衡步骤,净化效果显著,适合大规模样品的快速净化处理。
试验同步考察了Oasis PRIME HLB 填料用量 (1 mL 30 mg;3 mL 60 mg;3 mL 150 mg;6 mL 200 mg;6 mL 500 mg)对目标物的吸附净化效果。由图2b 可知,当填料由1 mL 30 mg 增到3 mL 200 mg 时,6 类动物肝脏的萃取效率迅速提高;当填料量为6 mL 200 mg 和6 mL 500 mg 时,两者对猪肝、牛肝和羊肝中药物的吸附净化效果相当,而前者对其它3 类禽类肝脏基质的吸附效果均最好。最佳填料量为6 mL 200 mg。
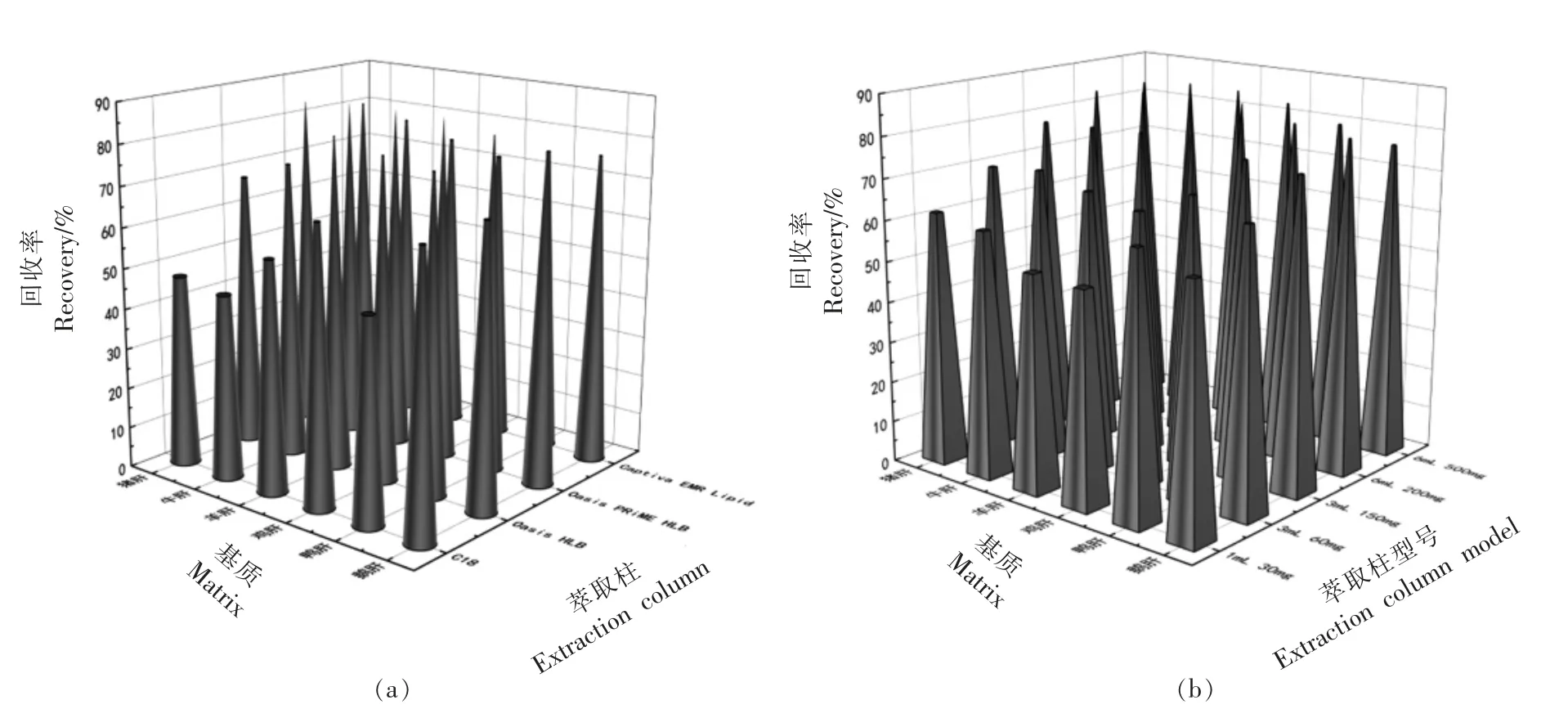
图2 不同萃取柱(a)及填料量(b)对45 种兽药回收率的影响Fig.2 Effects of extraction column (a) and packing amount (b) on recovery of 45 veterinary drugs
2.1.3 针式滤膜 针式滤膜可将提取与净化后的测试液中悬浮的颗粒物质充分过滤。以过滤后的样品与未过滤样品中化合物峰面积的比值作为评价依据,对0.22 μm 聚四氟乙烯(A)、0.22 μm 尼龙滤膜(B)和0.22 μm 聚醚砜(C)3 种滤膜的净化效果,结果发现,样品溶液经3 种材料的针式滤膜过滤后,均有大量化合物被检出,其中采用0.22 μm 聚四氟乙烯的检出化合物最多,说明其滤膜过滤净化效果最好。3 种滤膜过滤效果依次为0.22 μm 聚四氟乙烯>0.22 μm 尼龙滤膜>0.22 μm聚醚砜。为了最大程度减小兽药的损失,最终选用聚四氟乙烯材质滤膜(0.22 μm)过滤。
2.2 优化的UHPLC 条件
2.2.1 色谱柱 考察了Thermo Accucorea Q C18(2.1 mm×100 mm,2.6 μm)、Agilent Eclipse plus C18(3.0 mm×150 mm,1.8 μm)以及Waters Acquity BEH C18(2.1 mm×100 mm,1.7 μm)3 种主流色谱柱对45 种化合物的分离效果。结果发现,相当部分的兽药在Thermo Accucorea Q C18色谱柱上出现峰型拖尾现象,Agilent Eclipse plus C18色谱柱虽对大部分兽药能有效分离,但部分大环内酯药物存在峰型尖锐度较差的现象。而Waters Acquity BEH C18色谱柱能较好地保留45 种化合物,峰型尖锐对称且无明显拖尾现象,这可能与该色谱柱独特的键合与极性基团端基封尾技术确保高兼容的保留性能有关。最终选择Waters Acquity BEH C18色谱柱。
2.2.2 流动相体系及梯度洗脱程序 流动相体系直接影响分析物的保留时间和峰型,由于45 种药物均易溶于乙腈,因此分别选取乙腈-水、乙腈-5 mmol/L 乙酸铵、0.1%甲酸-乙腈-5 mmol/L(20 mmol/L)乙酸铵等4 种流动相组合进行质谱分析。结果发现,乙腈-水作为流动相时,各组分的峰型较差且正离子响应较低;乙腈-5 mmol/L 乙酸铵所获色谱峰型明显优于乙腈-水,部分四环素和青霉素类药物峰型较差。当乙酸铵浓度增至20 mmol/L时,峰型均得到明显改善,继续向乙腈-20 mmol/L乙酸铵溶液中添加0.1%甲酸,此时各待测物峰型均达到最佳,表明适量加入甲酸有利于改善峰型。本试验流动相体系:乙腈-20 mmol/L 乙酸铵(0.1%甲酸)水溶液。
采用梯度洗脱以实现完好分离。首先将初始流动相设置为98%,使极性大的物质先出峰,随后降低乙腈的起始浓度并保持浓度缓慢增加,以分散各组分的出峰时间,确保流动相的极性在一个合适的范围变化。随着极性的变化,以高浓度乙腈保持一段时间,使不同极性范围的化合物依次出峰,避免色谱峰重叠,然后将乙腈浓度降至初始浓度水平并保持平衡。综合考虑色谱峰的峰型、灵敏度、出峰时间等因素,梯度洗脱程序设置见表1。
2.3 优化的HRMS 条件
45 种化合物在HESI 电离源下产生多种准分子离子峰,采用全扫描/数据依赖二级扫描(Full MS/dd-MS2)模式对45 种药物混合标准溶液进行正负离子扫描,得到一级全扫描质谱图。以各化合物的精确质量数提取色谱图,此条件下45 种化合物的精确质量相对偏差均小于1 ppm,满足试验需求。同时,在多种分辨率考察中发现,当一级全扫描质量分辨率为75 000,数据依赖二级扫描质量分辨率为17 500 时,所有待测物与样品基质中的干扰物均实现基线分离,响应值得到明显提高,表明在此分辨率条件下样品中的基质干扰得到有效消减。图3为45 种药物提取的离子色谱图。


图3 45 种兽药化合物提取的离子色谱图Fig.3 Extraction ion chromatograms of 45 veterinary drug compounds
2.4 基质效应评价结果
动物肝脏基质十分复杂,存在基质抑制或增强效应的可能性。按照基质效应计算方法,对猪肝、牛肝、羊肝、鸡肝、鸭肝和鹅肝等6 种样品开展基质效应评价。结果表明,猪肝、牛肝、羊肝、鸡肝、鸭肝和鹅肝均存在基质效应。其中,猪肝和鸡肝表现为强基质效应的药物占比较高。为降低基质效应,获得更加准确的检测结果,在定量环节通过基质匹配标准曲线来消除或减弱基质效应的影响。
2.5 方法的线性关系、检出限和定量限
将6 种动物肝脏空白提取液配成45 种化合物的标准工作液,在优化的分析条件下测定。以目标化合物色谱峰面积(y)及其对应组分浓度(x)绘制标准曲线,计算获得待测物质的线性回归方程及其相关系数(r)。分别以3 倍信噪比(S/N=3)和10 倍信噪比(S/N=10)计算方法的检出限(Limit of detection,LOD) 和定量限 (Limit of quantitation,LOQ)。结果表明,当质量浓度为1~2 000 μg/L 时,45 种化合物展现出良好的线性关系,所对应的相关系数r 均大于0.9959。方法的检出限和定量限依次为0.5~20 μg/kg 和2~50 μg/kg,均满足试验需求。表3列出猪肝典型样品中45 种药物的线性关系、检出限和定量限。
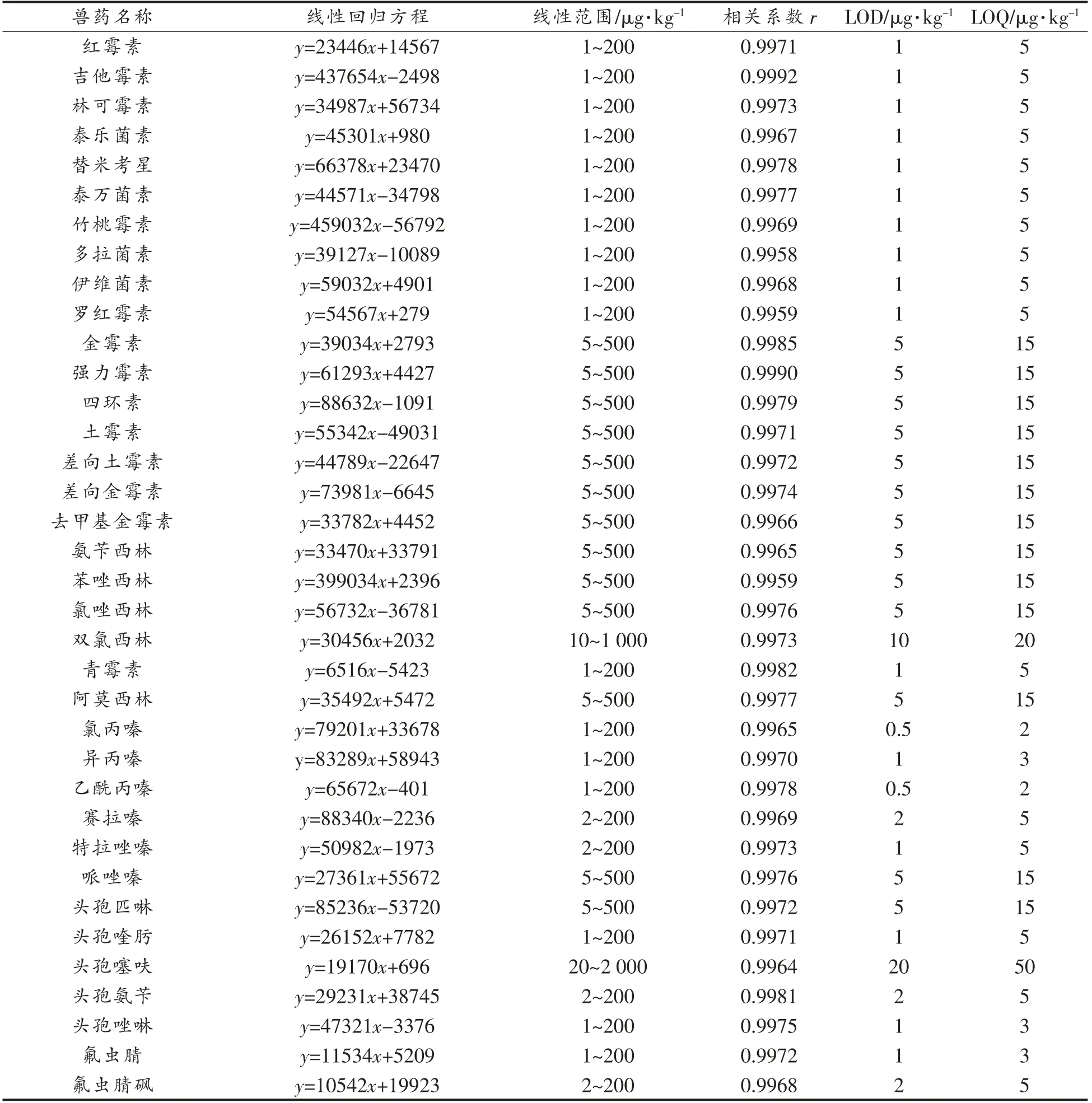
表3 猪肝中45 种兽药的线性关系、检出限和定量限Table 3 Linear relationships,limits of detection and limits of quantification of 45 veterinary drugs in pig liver

(续表3)
2.6 回收率和精密度
分别向6 种动物肝脏的空白样品中添加45种兽药化合物的标准溶液配成阳性样品,按照已建立的前处理和仪器分析方法,在1 倍LOQ、5 倍LOQ 和10 倍LOQ 3 个添加浓度水平下各重复测定6 次,求解得到回收率及相对标准偏差(Relative standard deviation,RSD)。结果显示,添加水平在1 倍LOQ~10 倍LOQ 时,45 种药物在猪肝中的回收率为76.5%~103.8%,RSD 为6.1%~9.6%;牛肝中的回收率为72.0%~102.5%,RSD 为5.7%~9.6%;羊肝中的回收率为79.8%~112.2%,RSD 为5.0%~10.3%;鸡肝中的回收率为72.5%~103.6%,RSD 为6.1%~9.5%;鸭肝中的回收率为78.5%~104.5%,RSD 为6.2%~10.5%;鹅肝中的回收率为83.6%~109.6%,RSD 为5.4%~9.3%。表明本研究建立的方法准确度和精密度均满足试验需求。表4为两种典型(猪肝、鸡肝)样品中45 种药物的回收率和精密度。
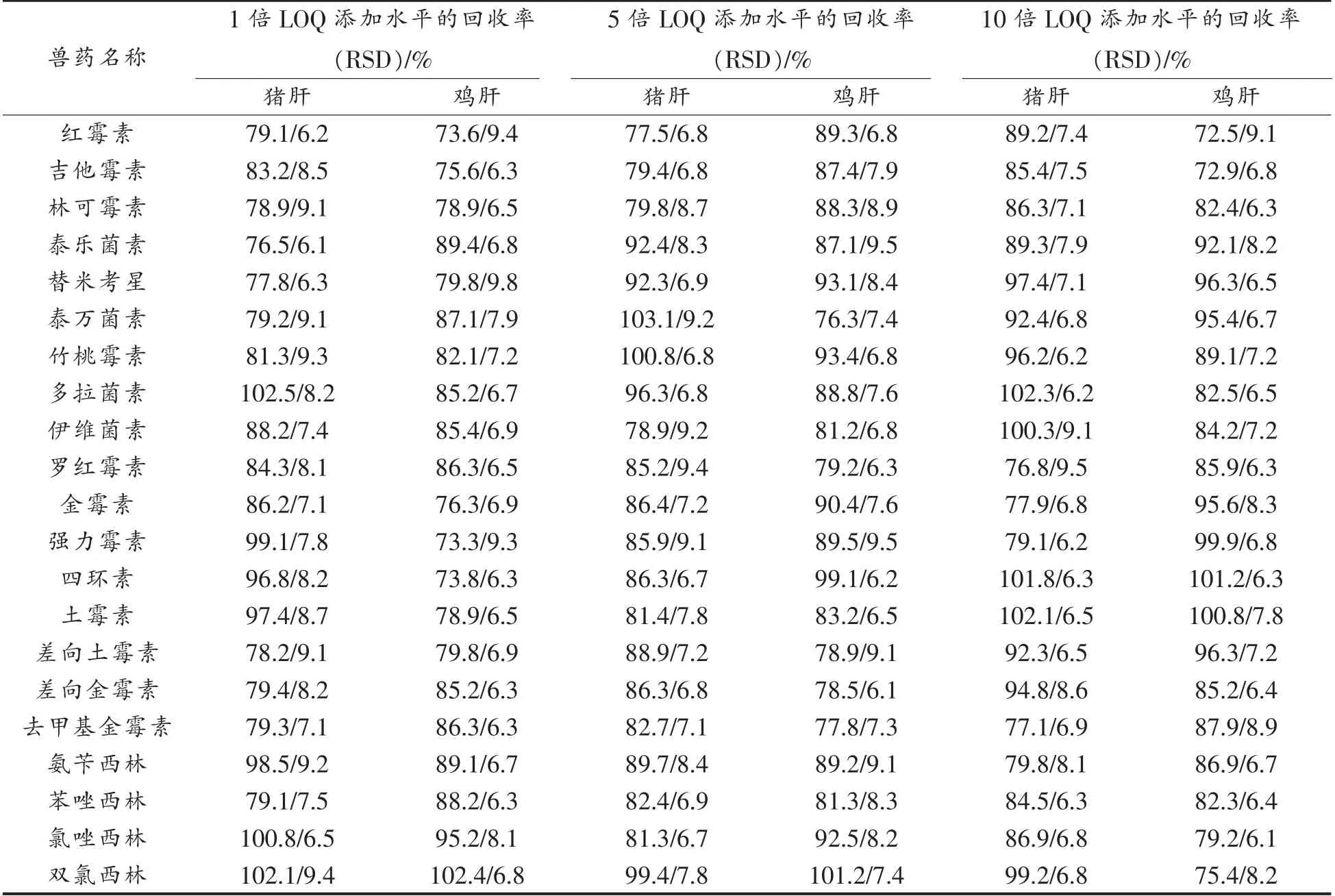
表4 45 种兽药在猪肝和鸡肝中的回收率和相对标准偏差(n=6)Table 4 Recoveries and relative standard deviations of 45 veterinary drugs in pig liver and chicken liver (n=6)
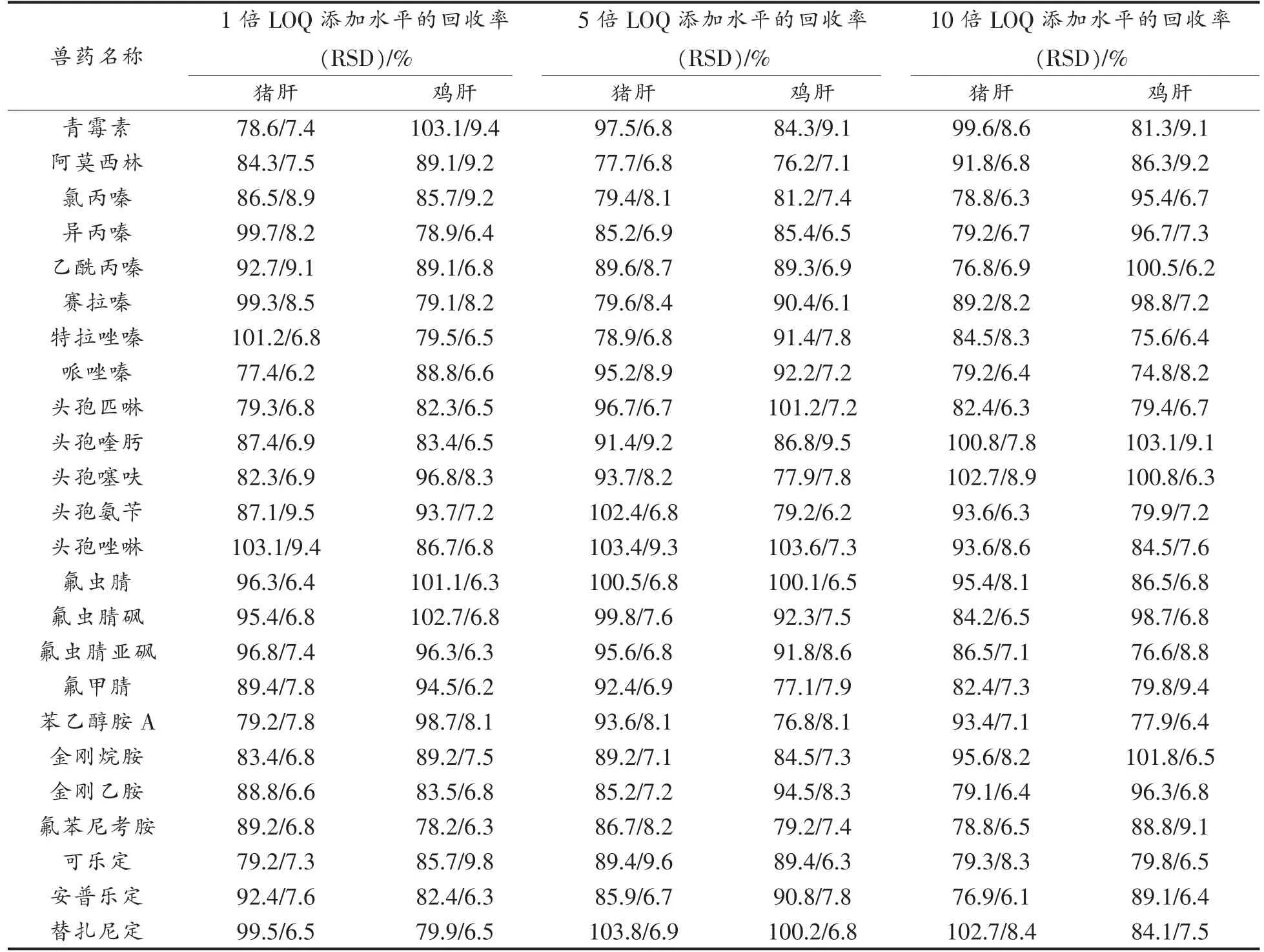
(续表4)
2.7 实际样品筛查结果
在市场上随机采取54 批次动物肝脏样品(猪肝、牛肝、羊肝、鸡肝、鸭肝、鹅肝各9 批次)进行检测,样品经前处理和上机测试后,利用所建数据库中色、质谱指纹识别信息与HCD 二级特征碎片离子对样品中45 种兽药进行多目标筛查与确证,使用基质匹配标准曲线定量。结果显示,在1 批次猪肝中检出少量残留的金霉素(6.7 μg/kg),符合国家限量标准值(≤100 μg/kg),其余样品均未检出45 种药物。
3 结论
在优化PRiME HLB 前处理条件的基础上,利用高分辨质谱技术建立的电子化数据库分析目标化合物,提高了非靶向多目标精准侦测能力。首次建立了快速筛查6 种动物肝脏复杂基质中45种兽药的UHPLC-Q-Orbitrap HRMS 方法。方法的前处理简单、通量高、准确可靠,为动物肝脏中兽药残留风险识别与隐患排查提供技术支持。
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
煤化工(2022年3期)2022-07-08
辽宁化工(2022年3期)2022-04-06
皮革制作与环保科技(2021年14期)2021-11-12
科学家(2021年24期)2021-04-25
辽宁化工(2021年1期)2021-02-22
首都食品与医药(2020年1期)2020-10-21
中国动物保健(2019年3期)2019-12-11
山东工业技术(2016年10期)2016-09-06
云南农业(2015年10期)2015-03-19
