
大豆分离蛋白-维生素D3复合物的结构及性质
2022-08-17 05:56石佳卉张安琪邵志远王喜波江连洲
中国食品学报 2022年7期
石佳卉,张安琪,陈 爽,邵志远,王喜波*,江连洲
(1 东北农业大学食品学院 哈尔滨 150030 2 黑龙江省农垦龙王食品有限责任公司 黑龙江绥化 152000)
维生素D 是脂溶性微量元素,在人体细胞生长、分化中起重要作用,可促进钙、磷吸收和转运,还与自身免疫性疾病有关[1-2]。在生活、饮食习惯及年龄等因素的影响下,维生素缺乏在全球范围内普遍存在,尤其是维生素D 缺乏[3-4]。维生素D3(Vitamin D3,VD3) 具有维生素D 最高的生物活性[5],然而,其在光、热条件下不稳定,容易在空气中氧化分解,失去活性[6]。
为了提高VD3的稳定性及其生物利用度,常采用糖类和蛋白质作为封装载体负载保护VD3[7-8]。理论上,作为纳米复合物载体,蛋白质比糖类更有优势[9]。大豆分离蛋白(Soy protein isolate,SPI)作为一种功能特性良好,营养价值丰富的植物蛋白,因无毒、廉价等优点,而被用于食品封装技术中。Chen 等[10]和Pujara[11]分别用SPI 对姜黄素和白藜芦醇进行封装,均有效提高了被封装物质的水溶性;Lee 等[12]用pH 偏移和超声结合制备可溶性SPI 纳米聚集体并对VD3进行封装,使其抵抗紫外线破坏的能力提高。
大豆蛋白通过静电作用和疏水相互作用与可电离或疏水性小分子结合,因此大豆蛋白和VD3互作是提高VD3稳定性的一种途径。目前关于SPI-VD3复合物结构性质的研究较少。本团队前期研究了不同均质压力、均质次数处理SPI-VD3复合物的结构性质[13-14]。本文选用不同浓度的VD3分别与SPI 复合,研究不同浓度VD3对复合物粒径、电位、浊度和表面疏水性的影响,阐明VD3与SPI 相互作用后SPI 荧光光谱、紫外光谱和傅里叶红外光谱的变化规律及其二级结构变化,旨在为营养强化VD3技术及拓宽大豆蛋白的应用提供理论依据。
1 材料与方法
1.1 材料与试剂
维生素D3(Vitamin D3,VD3),纯度>99%,美国Sigma 公司;低温脱脂豆粕,山东禹王实业有限公司;ANS 荧光探针,美国Sigma 公司;浓盐酸,哈尔滨理工化学试剂有限公司;试验试剂均为分析纯级。
1.2 设备与仪器
GL-21M 型冷冻离心机,长沙湘智离心机仪器有限公司;ALPHA 1-4 LSC 型冷冻干燥机,德国Christ 公司;FTIR-8400S 型傅里叶红外光谱仪、UV-240IPC 型紫外分光光度计,日本岛津公司;F-4500 型荧光分光光度计,日本日立公司;Zetasizer Nano ZS90 型Zeta 粒度及电位测定仪,英国Malvern 公司。
1.3 试验方法
1.3.1 SPI 的制备 根据Sorgentini 等[15]的方法制备SPI,略作改动。取一定量低温脱脂豆粕粉碎,使用60 目筛筛分得到脱脂豆粉。用碱溶酸沉法提取SPI。为除去SPI 沉淀中的盐离子,将SPI 沉淀物用去离子水洗涤2 次。用2 mol/L NaOH 溶液调节SPI 溶液的pH 值至7.0。将样品倒入平皿后,于-20 ℃冰箱中预冻24 h,然后在-40 ℃下冷冻干燥。制备出的SPI 粉末储存于冰箱(-20 ℃)中备用。本试验提取的SPI 中蛋白含量为90.12%。
1.3.2 VD3溶液的制备 避光下称取一定量VD3粉末溶解在无水乙醇中,使用磁力搅拌使其充分溶于乙醇,制备好的VD3溶液避光储存在棕色瓶中备用。本研究中VD3溶液现用现配。
1.3.3 SPI-VD3溶液的制备 将一定量SPI 粉末加入磷酸盐缓冲液(0.01 mol/L,pH 7.0)中,配成SPI 溶液(4 mg/mL),按照SPI∶VD3的质量比为25∶1,20∶1,15∶1,10∶1,5∶1,将VD3溶液与SPI 溶液混合,室温(25 ℃)下避光搅拌1 h,各样品分别表示为SPI-VD3-1~SPI-VD3-5。
1.3.4 粒径测定 用粒度分析仪测定复合体系粒径。将SPI 溶液及SPI-VD3-1~SPI-VD3-5 样品溶液稀释4 倍。分散剂设定为水,分散剂和颗粒折射率分别为1.330 和1.460,颗粒吸收率设为0.1。
1.3.5 ζ-电位测定 采用Zetasizer Nano ZS90 型Zeta 电位测定仪分别测定SPI 溶液及SPI-VD3-1~SPI-VD3-5 样品的ζ-电位。测定前先用0.01 mol/L pH 7.0 磷酸盐缓冲液稀释待测样品,稀释至溶液蛋白质质量浓度为1 mg/mL,然后注入样品池。仪器操作温度设定为25 ℃,蛋白质和分散剂的折射率分别为1.450 和1.330,吸收率为0.001。
1.3.6 表面疏水性测定 参照Hayakawa 等[16]的方法测定表面疏水性。用磷酸盐缓冲液(0.01 mol/L,pH 7.0)稀释SPI 及样品溶液,使最终蛋白质量浓度为1,0.2,0.04,0.008 mg/mL。将20 μL 的ANS荧光探针(8 mmol/L,用pH 7.0,0.01 mol/L 磷酸盐缓冲液配制)与4 mL 样品均匀振荡,避光处理放置15 min。在激发波长、发射波长分别为390 nm和470 nm 下测定样品的荧光强度。以蛋白浓度为横坐标,荧光强度为纵坐标拟合曲线,直线斜率为表面疏水性。
1.3.7 浊度测定 参照Lee 等[12]的方法测定样品浊度。以不同蛋白VD3质量比的样品在600 nm 波长下测得的吸光值表示浊度,用蒸馏水作为空白。
1.3.8 荧光光谱测定 参照Liu 等[17]的方法测定荧光光谱。将SPI 溶液及SPI-VD3样品溶液稀释至蛋白质质量浓度为0.5 mg/mL,设定激发波长290 nm,在发射波长300~500 nm 范围内扫描,设定电压700 mV,狭缝宽度2.5 nm。
1.3.9 紫外-可见光谱分析 将SPI 溶液及不同蛋白VD3质量比的样品分别稀释至0.2 mg/mL,分别移取3 mL 于石英比色池中,吸收光谱扫描范围200~400 nm。
1.3.10 傅里叶红外光谱分析 将SPI 和SPI-VD3样品冻干备用。在干燥环境下,按KBr∶样品的质量比为150∶1 的比例压制固体薄片[18]。使用8400S FTIR 红外光谱仪对固体薄片进行扫描,扫描次数为32 次,扫描范围为4 000~400 cm-1,分辨率为4 cm-1。采用PeakFit 4.12 软件拟合分析红外光谱,得到SPI 及SPI-VD3样品中二级结构含量。
1.3.11 数据统计与分析 利用Origin 8.0 软件处理数据作图,每组数据均重复3 次。数据处理与方差分析(ANOVA)使用IBM SPSS Statistics 20进行分析,P<0.05 表示差异显著。
2 结果与分析
2.1 VD3 添加量对SPI-VD3 复合物粒径、PDI 和ζ-电位的影响
如图1所示,SPI 粒径呈现双峰分布,随着VD3的加入,SPI-VD3复合物的体积粒径逐渐朝着单峰分布转变,体积粒径小的峰占比增大,体积粒径较大的峰占比减小。由图2同样可以看出随着VD3含量增加,体系平均粒径由384.3 nm 明显减小到125.2 nm,且多分散性指数PDI 变小,说明体系中粒径分布更均一,稳定性较强。可能是由于VD3的存在,与SPI 通过疏水相互作用和氢键等自组装成为胶束,且该胶束的胶束结构比SPI 胶束更加紧密[19]。样品ζ-电位变化范围为-19.6~-20.3 mV,随着VD3含量的增多,电位的绝对值逐渐增大,当SPI 与VD3质量比为10∶1 时,电位绝对值最大,此时体系最稳定。也可能是由于VD3与SPI 交联后使SPI 溶液的ζ-电位绝对值升高,有助于保持溶液粒子之间彼此远离,从而导致溶液液滴尺寸减小[20]。

图1 SPI-VD3 复合物的ζ-电位和粒径分布图Fig.1 ζ-potential and particle size distribution of SPI-VD3 complex

图2 SPI-VD3 复合物的平均粒径和PDIFig.2 Average particle size and PDI of SPI-VD3 complex
2.2 VD3 添加量对SPI-VD3 复合物表面疏水性的影响
表面疏水性(H0) 与蛋白的功能性质密切相关。如图3所示,随着VD3浓度的增大,SPI-VD3复合物的表面疏水性显著降低 (P<0.05)。当SPI与VD3质量比为5∶1 时,表面疏水性由1 137.67降到了1 008.57。这表明添加VD3后,SPI 表面更加亲水,可能是VD3引起了SPI 结构改变。H0下降可能是由于VD3与SPI 相互作用,导致部分掩埋在SPI 内部的亲水基团暴露出来[21]。同时,VD3是疏水性物质,VD3与SPI 疏水基团相互作用,从而减少了ANS 与SPI 结合的数量,这可能是VD3使SPI 表面疏水性下降的原因之一。

图3 SPI-VD3 复合物的表面疏水性Fig.3 Surface hydrophobicity of the SPI-VD3 complex
2.3 VD3 添加量对SPI-VD3 复合物浊度的影响
浊度是悬浮物对光线的阻碍程度,能够反映蛋白聚集程度及聚集体大小[22],同时也直接影响溶液的外观。由图4可知,VD3含量的增加使VD3-SPI 复合物的浊度略有增加,这与前述的SPI-VD3复合物粒径变化不同,这可能由于部分VD3没与SPI 相互作用形成复合物而是游离于体系中所致。

图4 SPI-VD3 复合物的浊度Fig.4 Turbidity of SPI-VD3 complex
2.4 VD3 添加量对SPI-VD3 复合物荧光光谱的影响
SPI 和SPI-VD3复合物的荧光发射光谱见图5。激发波长为290 nm 时,SPI 的荧光发射峰在360 nm 附近。随VD3比例的升高,荧光强度降低,表明VD3与SPI 之间存在相互作用,VD3起到猝灭剂作用,且浓度越大猝灭效果越明显。同时,荧光发射峰由360.8 nm 红移至364.4 nm,红移了3.6 nm。荧光发射峰的变化反映了蛋白中色氨酸残基和酪氨酸残基微环境的改变。微环境向亲水转变,说明SPI 与VD3结合,使SPI 空间结构发生变化,肽链结构展开[23]。

图5 SPI-VD3 复合物的荧光光谱Fig.5 Fluorescence spectra of SPI-VD3 complex
2.5 VD3 添加量对SPI-VD3 复合物紫外-可见吸收光谱的影响
在不同浓度VD3影响下SPI 的紫外-可见吸收光谱变化见图6。随着VD3浓度的增加,SPIVD3复合物的吸光度逐渐增强,最大吸收峰波长(λmax)由259 nm 红移至267 nm。λmax红移说明SPI中芳香族氨基酸的微环境发生变化,SPI 构象改变,肽链伸展,进而增强了吸光度。该结果与付彩霞等[24]关于VE 与牛血清蛋白的相互作用的研究结果一致。
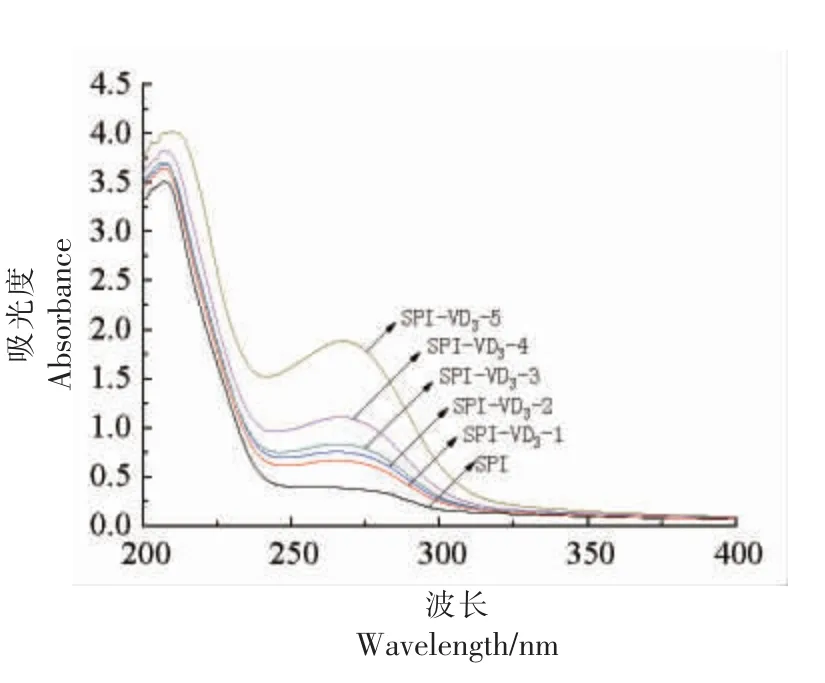
图6 SPI-VD3 复合物的紫外-可见吸收光谱Fig.6 Ultraviolet-visible absorption spectrum of SPI-VD3 complex
2.6 VD3 添加量对SPI-VD3 复合物傅里叶变换红外光谱的影响
由图7可知,添加VD3后酰胺Ⅰ区和酰胺Ⅱ区的透光率降低了,SPI 酰胺Ⅰ区从1 632.93 cm-1蓝移至1 631.96 cm-1,酰胺Ⅱ区从1 531.28 cm-1红移至1 533.13 cm-1,说明添加VD3使SPI 的二级结构发生了变化。

图7 SPI-VD3 复合物的傅里叶红外光谱Fig.7 Fourier transform infrared spectroscopy of SPI-VD3 complex
傅里叶红外变换光谱能够敏感地反映肽链结构的变化,得到的红外光谱图利用PeakFit 4.12软件进行处理,通过进行区域选定(1 600~1 700 cm-1)、基线校准,Savitsk-Golay 平滑处理,以及二阶导数的拟合处理得到蛋白质二级结构含量。酰胺I 区中频率范围1 648~1 664 cm-1被指定为α-螺旋结构,频率范围1 615~1 637 cm-1和1 682~1 700 cm-1被指定为β-折叠结构,频率范围1 664~1 681 cm-1和1 637~1 648 cm-1分别被指定为β-转角和无规卷曲结构[25]。从图8可以看出,与SPI 相比,α-螺旋和β-折叠含量下降,β-转角和无规则卷曲含量明显升高(P<0.05),这可能是由于VD3与蛋白质内部的疏水性氨基酸区域结合从而使蛋白质分子结构展开,α-螺旋和β-折叠转换为β-转角和无规则卷曲。α-螺旋的结构以紧密、坚固为特点,可能阻碍了SPI 某些功能特性的发挥。β-转角和无规则卷曲构象的增加,明显增强了蛋白质的柔韧性,对SPI 功能特性的发挥产生有利影响[26]。刘英杰等[27]研究花青素与SPI 的不同交联法对蛋白质结构的影响,发现酶法和碱法共价法均使SPI 结构中的α-螺旋和β-折叠含量降低,使SPI 肽链解折叠。Liu 等[28]研究发现,绿原酸、没食子酸与乳铁蛋白结合,使其α-螺旋减少,蛋白结构更加延展。

图8 SPI-VD3 复合物中蛋白的二级结构含量Fig.8 Content of the secondary structure of proteins in the complex of SPI-VD3
3 结论
1)VD3与SPI 相互作用后会降低SPI 的表面疏水性;VD3的添加使SPI 的粒径减小,ζ-电位的绝对值增大,使体系粒径分布更加均匀,溶液的稳定性增强;随着VD3含量的增加,复合物的浊度略有增大。
2)VD3使芳香族氨基酸残基所处的微环境向极性增强方向改变,SPI 分子构象发生变化,结构更加舒展;傅里叶红外光谱结果显示,VD3的加入引起大豆分离蛋白二级结构的变化,进一步说明VD3与SPI 发生了相互作用。
猜你喜欢
自动化仪表(2022年11期)2022-11-24
山西化工(2022年6期)2022-10-09
九江学院学报(自然科学版)(2022年2期)2022-07-02
药学与临床研究(2021年3期)2021-07-13
西华大学学报(自然科学版)(2020年6期)2020-10-15
绿色科技(2018年20期)2018-12-19
意林·全彩Color(2018年9期)2018-10-12
安徽化工(2018年4期)2018-09-03
筑路机械与施工机械化(2017年6期)2017-07-10
药学研究(2015年11期)2015-12-19
