
DNA甲基化转移酶hsdM基因对维氏气单胞菌运动性的影响
2022-08-16 00:45唐燕琼唐鸿倩
生物学杂志 2022年4期
陈 婷,李 宏,王 丹,唐燕琼,唐鸿倩,马 香,刘 柱
(海南大学 生命科学与药学院 热带生物资源教育部重点实验室,海口 570228)
DNA甲基化是一种常见的遗传修饰现象,在生物有机体中普遍存在,已被证实参与调控多种生命活动途径[1]。在真核生物中,胞嘧啶甲基化调节发育基因的表达,DNA甲基化异常会导致包括癌症在内的多种疾病[2-3];在原核生物中,DNA甲基化参与多个细胞过程,包括防止外来DNA入侵、细胞周期调控、DNA错配修复和基因表达调控[4]。在细菌基因组中存在3种不同形式的DNA甲基化:N6-甲基腺嘌呤(6mA)[5]、N4-甲基胞嘧啶(4mC)[6]和5-甲基胞嘧啶(5mC)[7],其中6mA是原核生物中最普遍存在的一种甲基化形式。
DNA甲基化的发生机制是通过位点特异性的DNA甲基转移酶(MTase),将S-腺苷甲硫氨酸(S-adenosylmethionine,SAM)中的甲基基团添加到腺嘌呤或者胞嘧啶[8]。MTase一般在限制修饰(RM)系统中与同源限制性内切酶一起形成蛋白复合物,该复合物在进入细胞后会切割外来DNA,同时在宿主DNA的相同识别位点处进行甲基化,以保护宿主染色体免于被裂解[9-11]。I型RM系统由限制性内切酶(HsdR)、修饰性MTase(HsdM)和特异性亚基(HsdS)组成,该限制修饰系统中MTase催化甲基从SAM转移到腺嘌呤的环外氨基,形成6mA修饰[12]。研究表明,限制修饰系统介导的DNA甲基化在转录水平上直接或间接地调节参与细菌黏附、定殖的毒力基因表达[13]。还有一类MTase没有同源的限制性内切酶,作为孤儿甲基转移酶独立发挥作用[14]。细菌中研究最多的两个孤儿MTase是大肠杆菌DNA腺苷甲基转移酶(Dam),以及新月形杆菌参与细胞周期调节的甲基转移酶(CcrM)[15-16]。研究表明,Dam和CcrM催化的位点特异性DNA甲基化参与DNA错配修复[17],细胞周期进程控制[18],染色体复制起始时间调节和基因表达调控[19-20]。鞭毛运动使得细菌能够从不利的环境向营养物质更丰富、更有利于细菌生存的环境移动,增加细菌的竞争能力[21-23],是细菌定殖宿主进行致病的关键因素[24]。最新研究表明,MTase参与细菌毒力和运动性的调节[25-26]。
维氏气单胞菌(Aeromonasveronii)隶属于弧菌科气单胞菌属,运动型嗜温气单胞菌类[27-28],是一种革兰氏阴性兼性厌氧菌,具有较强的环境适应能力,广泛存在于湖泊、河口、海洋等水生环境中,在液体和半固体条件下都有较强的运动能力和定殖能力。近年来,国内外越来越多的报道表明:维氏气单胞菌已经成为一种重要的人、鱼共患致病病原菌,不仅能感染鱼类,给水产养殖造成巨大的经济损失,也可以感染包括人在内的哺乳动物,对人类健康造成严重威胁[29]。通过对维氏气单胞菌C4基因组的分析,发现一个I型RM系统甲基转移酶基因hsdM,为探究该基因介导的6mA甲基化对维氏气单胞菌C4运动性的影响,研究构建敲除hsdM基因的维氏气单胞菌C4突变株,并通过细菌生长曲线、运动能力测定和相对差异表达基因检测,初步揭示DNA甲基转移酶调节细菌运动性的分子机制,为进一步探究维氏气单胞菌毒力和致病性的调控机制奠定基础。
1 材料与方法
1.1 材料
1.1.1 质粒、菌株及引物
自杀质粒pRE112、大肠杆菌WM3064菌株(Escherichiacoli,WM3064)、维氏气单胞菌C4(Aeromonasveronii,C4)均由本实验室保存。研究用于PCR扩增及阳性转化子验证的引物及其序列信息如表1所示,引物合成及测序均由上海生工生物工程股份有限公司完成。

表1 实验使用的引物Table 1 Primers used in the present study
1.1.2 试剂盒、酶及药品
细菌总RNA快速抽提试剂盒购自上海生工生物工程有限公司;2×PCR Mix、细菌基因组DNA提取试剂盒、质粒提取试剂盒、DNA/胶纯化试剂盒、无缝克隆连接酶2×ClonExpress Mix及HiScript II Q RT SuperMix for qPCR (+gDNA wiper)试剂盒均购自南京诺唯赞生物科技有限公司;KpnⅠ-HF、SacⅠ-HF限制性内切酶购自北京NEB公司;氨苄青霉素(Ampicillin,Amp)、氯霉素(Chloramphenicol,Chl)、二氨基庚二酸(2,6-Diaminopimelic Acid,Dap)及琼脂粉购自索莱宝科技有限公司;蛋白胨、酵母提取物及氯化钠均购自西陇科学股份有限公司。
1.2 方法
1.2.1 敲除菌株ΔhsdM构建
敲除菌株基于同源重组双交换的原理进行构建,过程如图1所示,概括如下:设计特异引物(表1)用于通过PCR扩增hsdM基因的上、下游同源片段;用限制性内切酶KpnⅠ-HF、SacⅠ-HF双酶切pRE112质粒得到线性化载体;利用无缝克隆连接酶2×ClonExpress Mix将上下游同源片段连接到线性化载体上,得到hsdM基因敲除重组质粒pRE112-hsdM;通过电激转化法将重组质粒转入大肠杆菌WM3064感受态细胞;采用双亲结合法,使得重组质粒从大肠杆菌WM3064穿梭到野生型维氏气单胞菌C4中;最后,在20%蔗糖板上筛选得到敲除菌株ΔhsdM。
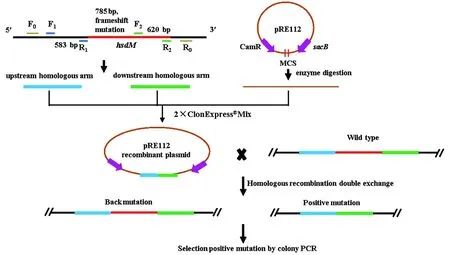
图1 维氏气单胞菌中基因hsdM敲除设计示意图Figure 1 Design diagram of hsdM gene deletion in A. veronii
1.2.2 生长曲线测定
从平板上挑取维氏气单胞菌C4野生型和ΔhsdM菌株的单菌落,将其分别接种到添加50 μg/mL氨苄青霉素(Amp)的LB液体培养基中,在30 ℃条件下150 r/min振荡过夜培养;次日将菌液以0.02 OD600/mL为终浓度转接到含50 μg/mL Amp的新鲜LB液体培养基中,在30 ℃条件下150 r/min振荡培养,之后每隔2 h取样,分别测定培养物的OD600数值,并绘制野生型和ΔhsdM菌株的生长曲线。
1.2.3 细菌运动性实验
挑取维氏气单胞菌C4野生型和ΔhsdM菌株的单菌落,分别接种于含有50 μg/mL Amp的LB液体培养基中,于30 ℃以 150 r/min振荡培养12 h;将菌液以0.02 OD600/mL为终浓度转接到含50 μg/mL Amp的LB液体培养基中,于30 ℃以150 r/min振荡培养20 h,测定OD600值。取1 mL菌液,用PBS(10 mmol/L,pH 7.4)洗涤一遍后,用PBS 稀释100倍,将1 μL稀释后的菌液接种于含有0.3%琼脂的LB固体培养基上,将平板室温放置20 min后,置于30 ℃培养箱培养4 h,观察实验结果。
1.2.4 实时荧光定量PCR
将维氏气单胞菌C4野生型和ΔhsdM菌株培养至稳定期后,收集菌体,使用细菌总RNA快速抽提试剂盒(Bacteria Total RNA Isolation Kit B518625)提取细菌总RNA。使用HiScript II Q RT SuperMix for qPCR (+gDNA wiper)试剂盒去除残留的基因组DNA污染,将RNA反转录为cDNA。用罗氏 LightCycler 96实时荧光定量PCR仪进行RT-qPCR实验。反应结束,采取2-ΔΔCt法分析实验结果,并用Graphpad Prism 8绘制柱状图。
2 结果与分析
2.1 敲除菌株ΔhsdM构建
使用pRE112载体验证引物,对转化hsdM基因上下游同源臂片段与pRE112质粒连接产物的大肠杆菌WM3064转化子进行菌落PCR扩增验证,结果如图2所示:空载pRE112质粒为模板,获得PCR扩增产物长度约为500 bp;所筛选阳性转化子为模板,获得PCR扩增产物约1 700 bp,证明hsdM基因上下游同源臂与载体连接,重组质粒pRE112-ΔhsdM构建成功。

M:DL5000 DNA Marker;1~14:pRE112-ΔhsdM基因敲除重组质粒菌落PCR产物;15:阳性对照;16:阴性对照 。图2 菌落PCR验证hsdM基因敲除重组质粒pRE112-ΔhsdMFigure 2 Identification of recombinant plasmid pRE112-ΔhsdM using colony PCR
通过双亲接合将重组载体pRE112-ΔhsdM从大肠杆菌WM3064转移至野生型维氏气单胞菌C4,并在含20%蔗糖的Amp抗性LB固体培养基上筛选突变菌株。使用hsdM敲除验证引物对随机挑选的单菌落进行菌落PCR扩增,结果如图3所示:以野生型维氏气单胞菌C4基因组为模板的PCR产物约2 400 bp,而阳性突变株的菌落PCR扩增产物约1 600 bp,表明目标基因已经从基因组敲除。挑选3个阳性突变株的PCR产物进行回收并送测序,测序结果显示ΔhsdM菌株构建成功。
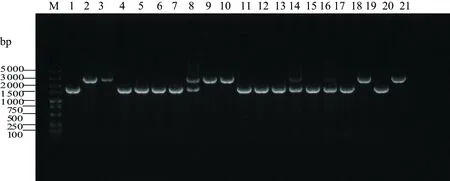
M:DL5000 DNA Marker;1~19:hsdM敲除株菌落PCR产物;20:阳性对照;21:阴性对照。图3 菌落PCR验证维氏气单胞菌hsdM敲除株Figure 3 Identification of A.veroniis train with hsdM deletion using colony PCR
2.2 野生型和ΔhsdM生长曲线检测
为探究hsdM基因敲除对维氏气单胞菌生长的影响,检测LB培养基条件下维氏气单胞菌C4野生型和ΔhsdM的生长曲线。结果显示(图4):生长期的前6 h,敲除株与野生型的生长速率基本一致;从8 h开始,敲除株生长速率略低于野生型,配对样品t检验显示二者生长能力存在显著差异(P<0.01);16 h后,野生型生长进入稳定期,敲除株还在缓慢生长,直至与野生型生长水平趋于一致。
2.3 野生型和ΔhsdM的运动性检测
通过平板运动实验探究hsdM基因敲除对维氏气单胞菌运动能力的影响,结果如图4所示:野生型和ΔhsdM菌株在平板上形成的菌落形态差异明显,野生型向四周发生显著迁移,且还有继续移动的趋势;而ΔhsdM菌株迁移距离明显较小[图5(a)]。根据两株菌的迁移距离绘制柱状图,对数据进行配对t检验发现二者间具有极显著差异[图5(b)]。结果表明,hsdM基因敲除极显著限制了维氏气单胞菌的运动能力。
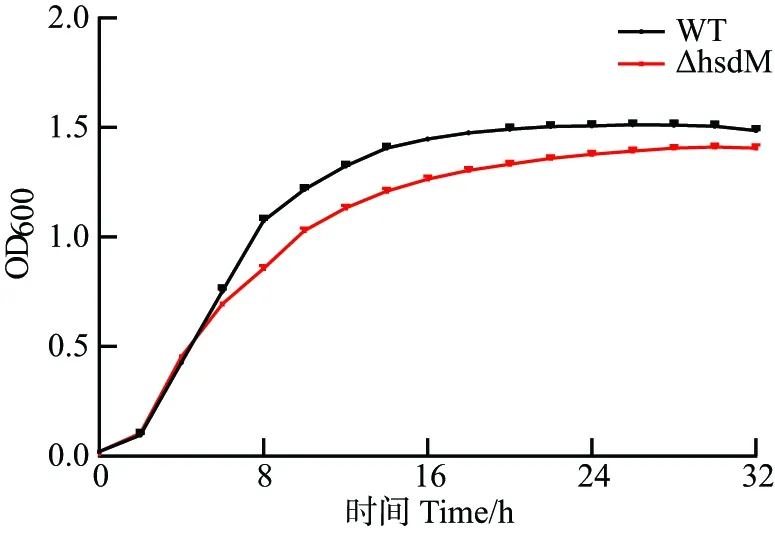
采用配对样品t检验确定各时间点组间生长差异;n=3。图4 维氏气单胞菌C4野生型和hsdM敲除株生长曲线测定Figure 4 Growth curves of WT and ΔhsdM

采用配对样品t检验确定组间迁移距离的差异;***P<0.001;n=3。图5 hsdM敲除对维氏气单胞菌C4运动能力的影响Figure 5 Effects of hsdM deletion on motility
2.4 野生型和ΔhsdM鞭毛基因表达差异检测
细菌运动性与鞭毛功能密切相关。通过RT-qPCR实验检测了一系列鞭毛生物合成基因在维氏气单胞菌野生型和hsdM敲除株中的相对表达水平。检测结果如图6所示:与野生型相比,ΔhsdM菌株中flgE、flgK、fliH、fliI、fliM和flhH等6个基因的转录水平显著下调。结果表明,HsdM甲基转移酶可以调控维氏气单胞菌C4鞭毛合成基因的表达。
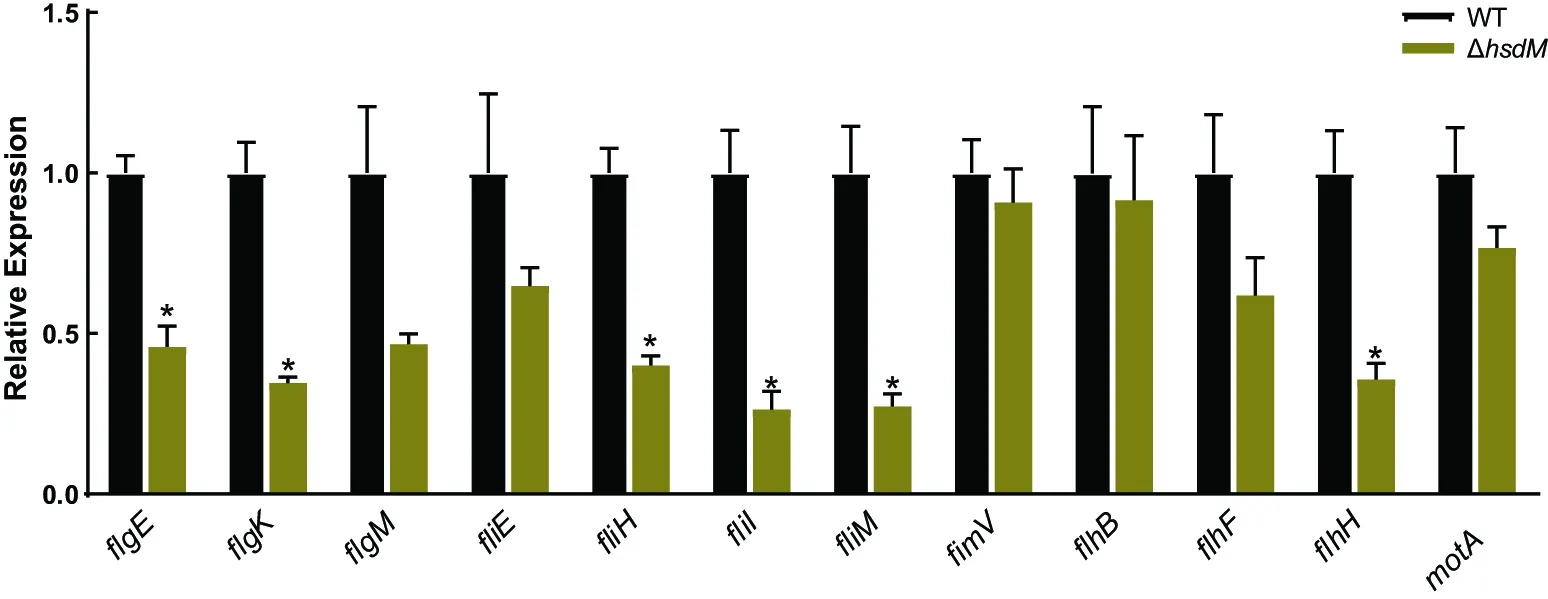
采用配对样品t检验确定组间鞭毛基因相对表达量的差异;* P<0.05;n=3。图6 维氏气单胞菌hsdM敲除株中鞭毛基因相对表达量变化Figure 6 Changes in relative expressions of flagellum genes caused by hsdM deletion in A. veronii
3 结论
研究初步确定I型RM系统甲基转移酶(MTase)基因hsdM缺失对维氏气单胞菌生长和运动能力的影响。与维氏气单胞菌野生型相比,ΔhsdM菌株的生长速率有微弱降低;运动能力极显著下降;同时,大部分鞭毛合成关键基因的表达水平下调。现有研究报道甲基转移酶是介导6mA甲基化的主要机制[13],甲基转移酶参与调节大肠杆菌的运动性和定殖[25],而且与结核杆菌毒力机制相关[26]。研究结果与上述报道一致,表明维氏气单胞菌中的HsdM甲基转移酶可能通过介导6mA甲基化而调控维氏气单胞菌C4鞭毛合成基因的表达,进而影响细菌的运动能力,甚至毒力机制。后续结合单分子实时测序(Single-molecule real-time sequencing,SMRT)技术绘制维氏气单胞菌的甲基化组[30],再通过甲基化分析和转录组测序分析hsdM参与调控细菌运动性的信号传导通路,有望进一步探明hsdM基因介导的6mA甲基化机制,及其对细菌运动性和毒力的调控机理。
猜你喜欢
保健与生活(2022年11期)2022-06-09
热带生物学报(2021年2期)2021-07-16
保健与生活(2021年5期)2021-04-12
陕西农业科学(2019年4期)2019-05-13
天然产物研究与开发(2018年10期)2018-11-06
环球时报(2018-10-24)2018-10-24
天然产物研究与开发(2018年8期)2018-09-10
中国农村科技(2018年11期)2018-01-17
冰雪运动(2016年6期)2016-05-17
三联生活周刊(2015年51期)2015-12-17
