
不同固化剂复配的耐高温环氧树脂体系性能
2022-08-11 02:36:02李伟捷光善仪徐洪耀
高分子材料科学与工程 2022年5期
李伟捷,光善仪,徐洪耀
(1.东华大学化学化工与生物工程学院生态纺织教育部重点实验室;2.东华大学分析测试中心及材料学院纤维改性国家重点实验室,上海 201620)
环氧树脂(EP)是指在特定条件下与固化剂发生开环交联反应,形成具有实际应用价值的三维网状结构的低分子量环氧化合物[1~3]。环氧树脂存在多种形式,常见的有液态、黏稠状和固体,未固化环氧树脂的使用价值并不高,只有在固化剂的作用下,反应生成了不溶不熔的三维交联聚合物,才具有优异的热性能、力学性能、耐化学性能,以及实际应用价值[4~6]。
固化剂的选择和复配是环氧树脂体系固化速率最重要的影响因素。近年来,国内外研究机构及材料厂商对环氧树脂的快速固化反应性进行了广泛的研究[7,8]。对于胺类固化剂而言,从脂肪胺到脂环胺再到芳香胺,其活性逐渐降低,固化速率也随之下降,但固化产物的力学性能逐渐升高[9~11]。美国氰特公司研究发现,不同活性的胺类固化剂复配使用存在“能量转移”的概念,即高活性脂肪胺在较低温度下开始反应,同时放出的热量引发低活性芳香胺反应,使整体反应速率大幅度加快,且反应产物性能优良[12,13]。目前对固化剂的研究主要集中在对其化学结构的改性,或固化剂的复配,而从复配固化剂的结构与含量对性能影响的研究较少。
因此,本文以环氧树脂E51 为基体树脂,分别将3 种不同结构的固化剂与芳香胺固化剂4,4’-二氨基二苯砜(DDS)复配,研究了不同固化剂及不同含量的同种固化剂对环氧树脂体系黏度、固化动力学及热性能的影响,旨在寻找一种复配固化剂,在降低体系黏度和反应活化能的同时,满足其他性能要求,阐述了复配固化剂结构对性能的影响并且为环氧树脂复合材料的制备提供理论依据。
1 实验部分
1.1 原材料
环氧树脂E51:工业级,深圳市吉田化工有限公司;DDS:分析纯,武汉远成共创科技有限公司;3,3’-二乙基-4,4’-二氨基二苯甲烷(DEDDM):工业级,菏泽永辉复合材料有限公司;4,4’-二氨基二苯甲烷(DDM):97%,罗 恩 试 剂;4-甲 基1,3-环 己 二 胺(HTDA):工业级,深圳市业旭实业有限公司。
1.2 样品制备
将脱模剂均匀涂抹在模具上,放入130 ℃的烘箱内烘干,以去除脱模剂中的溶剂和小分子。取一定量的环氧树脂E51 于三口烧瓶中加热升温,在130 ℃加入DDS,搅拌30 min 至DDS 完全溶解,冷却至80 ℃后分别加入3 种一定量的复配固化剂,搅拌均匀后抽真空除气泡30 min。而后将环氧树脂倒入涂有脱模剂的模具中,最后放入烘箱中90 ℃预固化1 h、130 ℃固化2 h、170 ℃固化3 h、200 ℃再后固化2 h。
1.3 测试与表征
1.3.1 黏度测试:采用标格达精密仪器(广州)有限公司的DV-Ⅱ型旋转式黏度计,选用31 号转子,称取约20 mL 树脂置于黏度计的试样筒中,预热3 min,逐渐升高温度,得到环氧树脂与固化剂混合后的变温黏度曲线。
1.3.2 差示扫描量热分析(DSC):采用德国耐驰公司的204 F1 型差示扫描量热仪测试环氧树脂的固化过程。取5~10 mg 已搅拌均匀的树脂混合物密封放入DSC 坩埚内,并以空坩埚作为空白样品,测试条件为N2气氛,分别以5 ℃/min,10 ℃/min,15 ℃/min 和20 ℃/min 的升温速率从50 ℃升温至350 ℃,得到不同升温速率下的固化曲线。
采用德国耐驰公司的204 F1 型差示扫描量热仪测试环氧树脂固化产物的玻璃化转变温度,测试气氛为N2,升温速率为10 ℃/min,采用升-降-升的形式消除热历史,测试温度范围为50~250 ℃。
1.3.3 动态力学热分析:采用瑞士梅特勒-托利多公司的动态力学热分析仪DMA1,样品尺寸为30 mm×7 mm×2 mm,选择三点弯曲模式,频率为1 Hz,升温速率为10 ℃/min,温度范围为50~300 ℃。
1.3.4 热失重分析:采用美国铂金埃尔默公司的TGA800 热重分析仪,N2气氛,升温速率为20 ℃/min,测试温度范围为50~600 ℃。
2 结果与讨论
2.1 黏度测试
黏度是环氧树脂复合材料加工过程中首先需要考虑的性能指标。Fig.2 分别为不同含量复配固化剂及不同种类复配固化剂对树脂体系黏度的影响。可以发现,随着温度的升高,不同复配固化剂树脂体系的黏度均逐渐减小,这是因为升高温度,分子链的活性增大,链段运动能力增强,体系的黏度减小;同时,在相同温度下,体系的黏度随着DDM 含量的增加呈现出减小的趋势,这是因为DDM 自身的熔点相较DDS 更低,与树脂混合相容性较好,可以有效降低体系的黏度,降低工艺加工温度。由右图不同复配固化剂对黏度减小的比率可以看出,HTDA 对黏度的降低效果最好,DEDDM对黏度的降低效果最差,这是因为HTDA 在常温时是一种黏度为8~15 mPa·s 的液体,而DEDDM 在常温时是一种黏度为2000~5000 的黏稠状液体,尽管都能有效地降低原有树脂体系的黏度,但是它们本身的黏度差异导致其对复合体系黏度降低的比率不同。右图中黏度降低的比率在100 ℃时有1 个拐点,这可能是因为在100℃时,少量的复配固化剂与环氧树脂发生反应,生成了一些低聚物,导致其与只加入DDS 的体系相比,黏度的降低发生迟滞,但当温度继续升高时,产生的低聚物溶解到环氧树脂中,使得体系相对黏度随温度升高的降低值减小,随着温度继续升高,相对黏度的降低又逐渐增大。

Fig. 1 Structure of curing agents

Fig.2 Viscosity-temperature curves and viscosity gradient of different resin systems
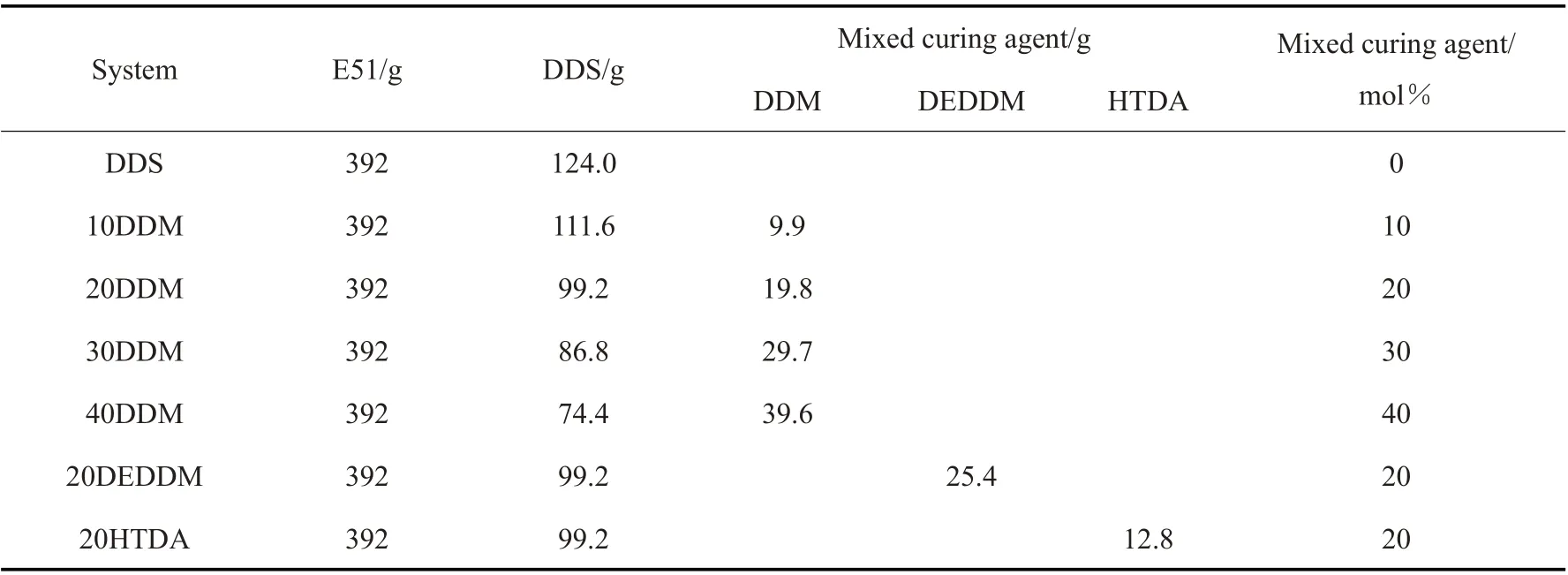
Tab.1 Formulation of compound curing agent resin systems

Fig.3 DSC curves of different contents of DDM at different heating rates
2.2 复配固化剂的不同配比对体系固化行为的影响
研究环氧树脂的固化反应动力学主要有2 种方法—等温法和非等温法。为了更好地了解环氧树脂在固化过程中的反应机理,获取更多固化过程中的信息,本文使用差示扫描量热仪,采用非等温DSC 法研究了不同升温速率下(5 K/min,10 K/min,15 K/min,20 K/min)环氧树脂的固化反应历程,并结合理论方程得到了反应活化能、反应级数和指前因子等一系列参数,构造了一个合理的方程来描述体系的固化反应历程[14]。
通过DSC 法研究反应动力学时遵循3 个基本假设:
(1)假设体系的固化反应速率与热流速率成正比
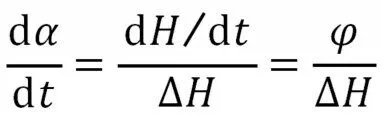
式中:da/dt——反应速率;φ——热流速率——固化体系总反应的放热晗。
(2) 假设体系的固化度为反应开始到t时的放热晗与总放热晗的比值
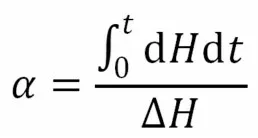
式中:α——固化度。
(3) 假设固化动力学分析的基本速率方程为
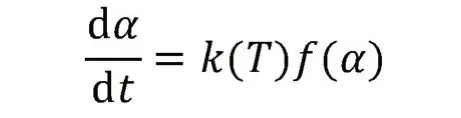
式中:K(T)——固化反应速率常数,并且遵循Arrhenius 方程,即
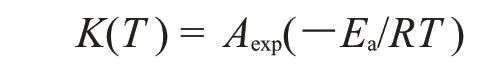
式中:T——绝对温度,K;Ea——反应活化能,kJ/mol;A——指前因子;R——气体常数。
所以,由以上假设可推出环氧树脂固化的动力学方程为
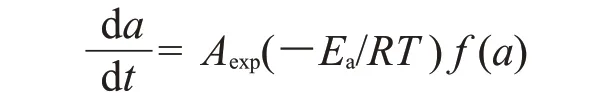
对于n级模型来说,可以通过Kissinger,Ozawa和Crane 方程来得到这些参数。
运用Kissinger 方程,假设峰值(Tp)处固化反应速率最快,即
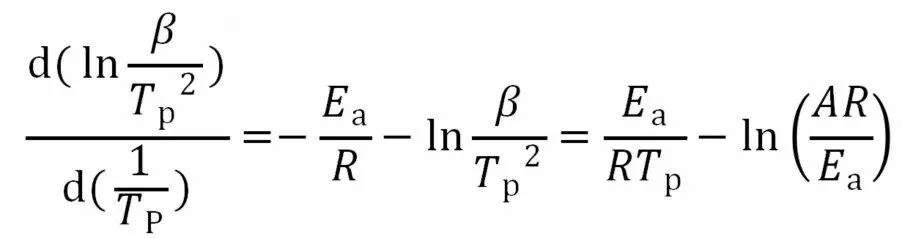
Ozawa 法
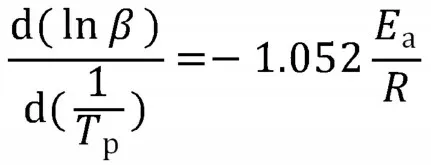
Crane 方程
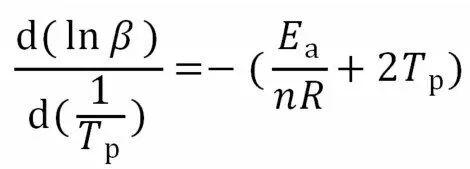
当Ea/nR>>2Tp有时,则
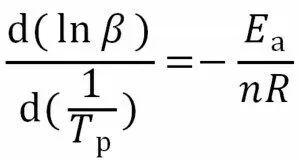
式中:β——升温速率,K/min;TP——峰值温度;R——理想气体常数,8.314 J/(mol·K);Ea——表观活化能,J/mol。
通过上文中的非等温DSC 曲线,可得Tab.2 中的各项参数。
通过对Tab. 2 中Ozawa 和Kissinger 方程参数进行线性拟合可得到Fig. 4。
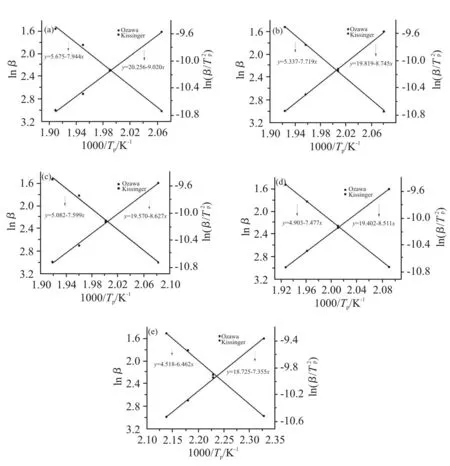
Fig.4 Kissinger and Ozawa fitting curves of different contents of DDM
由Kissinger 和Ozawa 方程拟合曲线的斜率通过公式计算得到Kissinger 活化能和Ozawa 活化能,并取平均值,再由Crane 方程计算得到反应级数(n),即可得n级反应动力学模型[15],即Tab.3 中的各项数据。
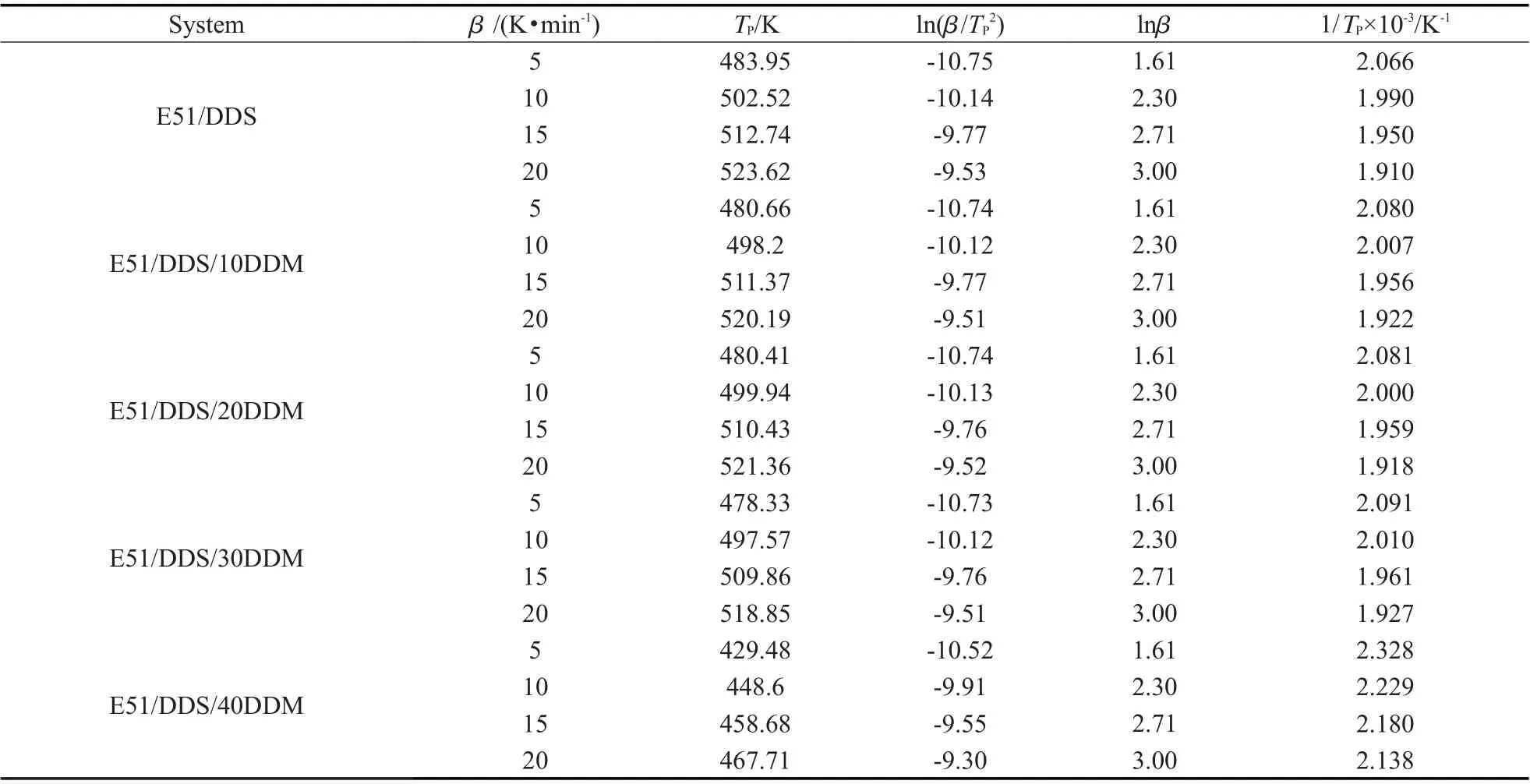
Tab. 2 Equation parameters of Kissinger method and Ozawa method for different systems
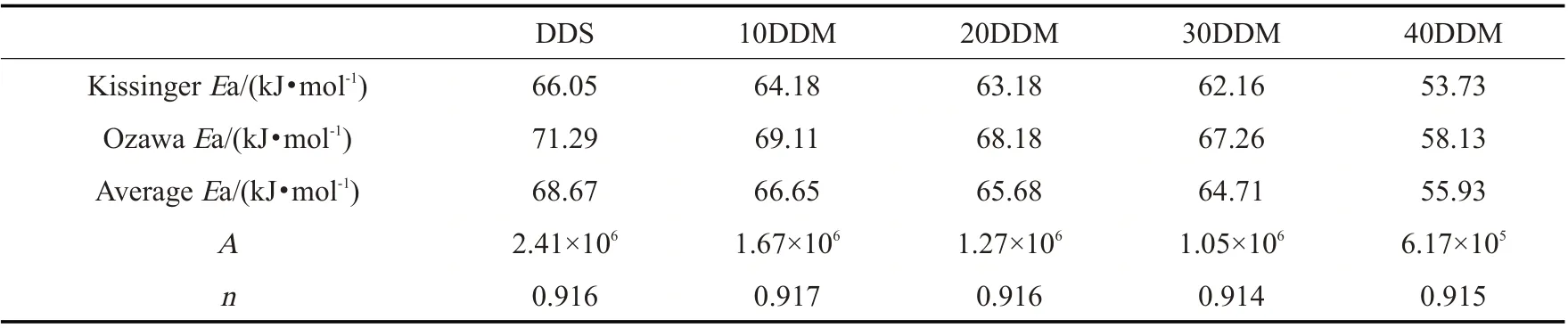
Tab.3 Kissinger activation energy, Ozawa activation energy and reaction order number n of different mixed curing agent systems
由Tab.3 各体系反应活化能的变化可以看出,随着DDM 加入量的增加,树脂体系的反应活化能随之降低,平均反应活化能从68.67 kJ/mol 降低至55.93 kJ/mol。这是因为DDS 中含有砜基,砜基作为一种吸电子基团,可使苯环的电子云密度降低,整个体系趋于稳定,两侧的氨基不易与环氧树脂中的环氧基团发生反应。并且,二苯砜基的化学键强度高,且整个二苯砜基处于高度共振状态,当吸收大量的热能和辐射能时,可以通过这种共振体系消散,而不发生链的断裂和交联。而DDM 与DDS 相比,2 个苯环中的砜基变为了亚甲基,导致DDM 两侧的氨基拥有更高的反应活性,更易致使环氧基团发生开环交联反应,因此,随着所含DDM 比例的提高,体系的平均反应活化能随之降低。
2.3 不同种类固化剂对体系固化行为的影响
Fig.5 为不同种类复配固化剂的非等温DSC 曲线。可以发现,不同升温速率下的固化曲线呈现单一的放热峰,这是因为体系中复配固化剂的占比为2:8,较低含量的固化剂在固化反应进行中放出的热量与DDS 固化时放出的热量相叠加,但是由于其占比较低,不足以产生另一个放热峰。随着升温速率的增加,固化放热峰向高温移动,这是因为在相同温度下,升温速率越快,固化度越低;达到相同的固化度时,升温速率越快,需要越高的温度,这种效应在整个升温过程中的积累导致了固化曲线向高温方向移动。
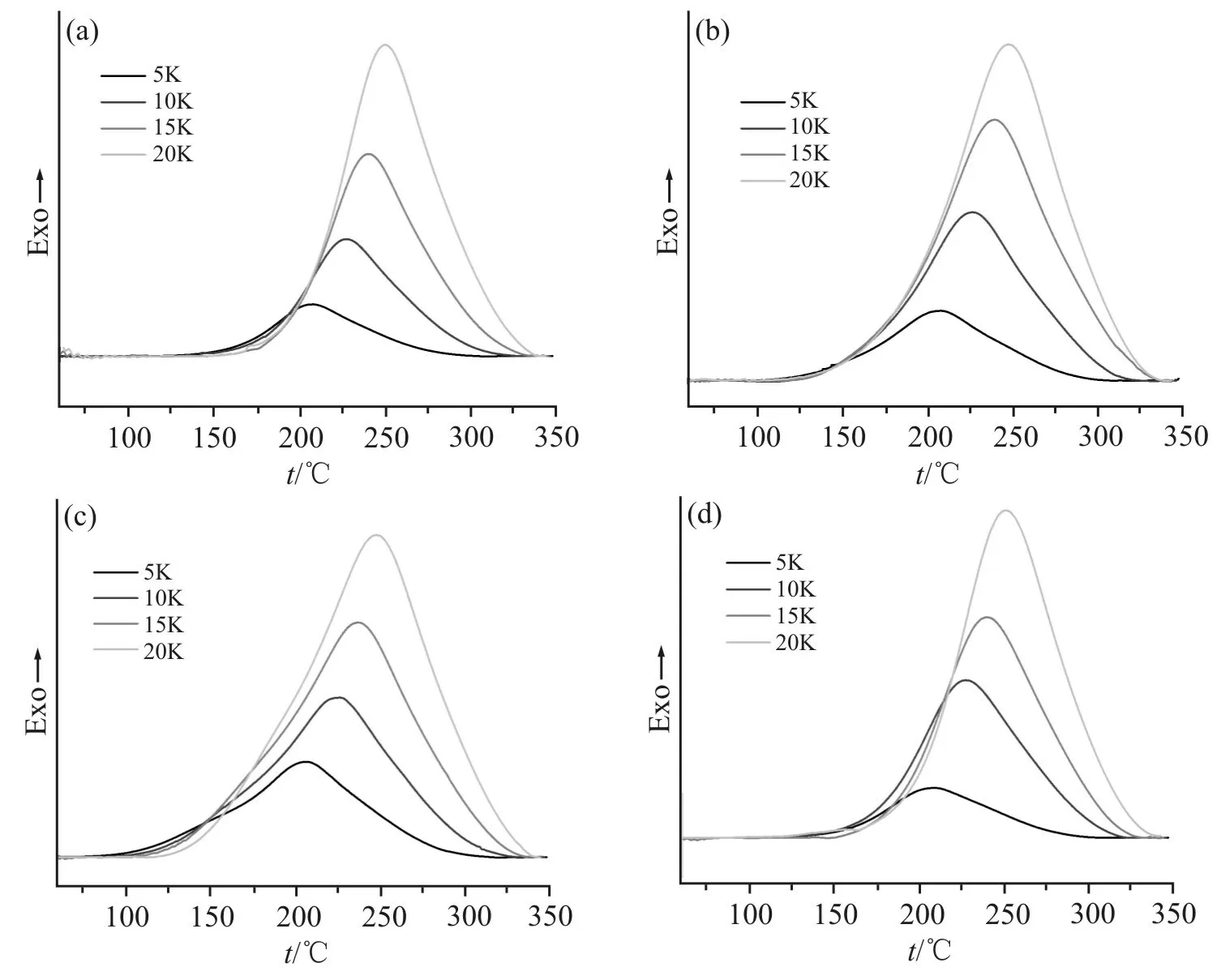
Fig.5 DSC curves of various systems at different heating rates
由Kissinger 和Ozawa 方程拟合曲线的斜率通过公式计算得到Kissinger 活化能和Ozawa 活化能,并取平均值,再由Crane 方程计算得到反应级数n,即可得n级反应动力学模型,即Tab.5 中的各项数据。
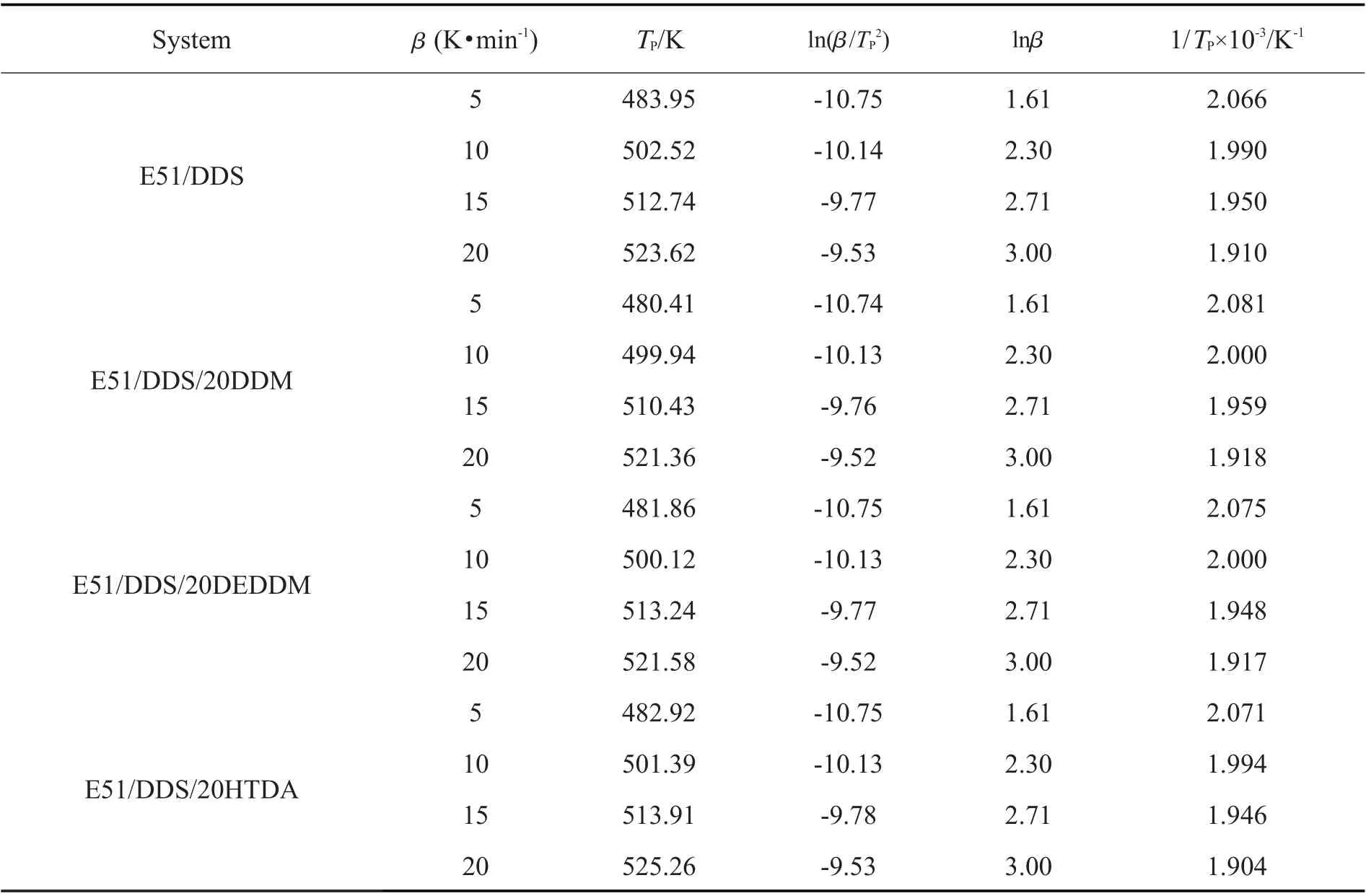
Tab. 4 Equation parameters of Kissinger and Ozawa method for different mixed curing agents systems
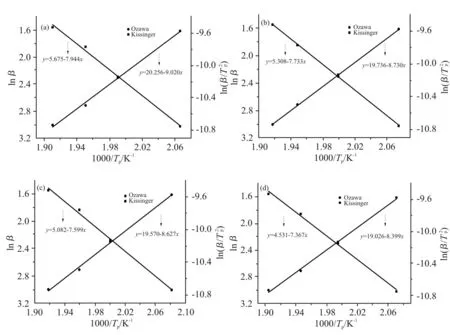
Fig.6 Kissinger and Ozawa fitting curves for different kinds of mixed curing agents
由Tab.5 中各体系反应活化能的变化可以看出,3 种复配固化剂体系的平均反应活化能相比纯DDS体系均有所降低。其中HTDA 的降低最为显著,从68.67 kJ/mol 降低至63.82 kJ/mol,降低了7.1%。这是因为对于二胺类的固化剂而言,其反应活性一般遵循脂肪胺>脂环胺>芳香胺,HTDA 作为其中唯一的脂环族胺类,其上的2 个氨基均与环己基相连,而DDS,DDM 和DEDDM 中氨基上的N 原子电子与苯环相互作用形成共轭体系而变得稳定,使得氨基的活性较低,需要更高的反应活化能。另一方面,DEDDM 对于反应活化能的降低最低,这是因为DEDDM 两侧的乙基有较大的空间位阻,对于环氧树脂交联体系的形成造成了阻碍,并且DEDDM 体系的黏度相较其余2 种较大,而较高的黏度会降低固化剂分子与环氧树脂分子间的碰撞频率,但是相较于DDS 中砜基对于苯环上所连氨基的共轭作用相比,这2 种效应的影响较小。因此,DEDDM 的加入能够降低体系的平均反应活化能,但是其对于平均反应活化能的降低效果较差,这与Tab.5 中反应峰值温度的规律是一致的,也进一步支持了活化能结果的可信度。
2.4 不同复配固化剂体系的玻璃化转变温度
Fig.7(a)为不同复配固化剂体系固化产物的DSC 图。由图可知,各体系均表现出1 个玻璃化转变温度,这说明复配固化剂体系与环氧树脂的相容性较好。从Fig.7(a)中可以看出,随着DDM 含量的增加,体系的玻璃化转变温度呈现下降的趋势,从227 ℃降低到207 ℃,下降了8.8%,这是因为DDM不含DDS 中具有强吸电子效应的砜基,导致整个体系的刚性下降,但是DDM 中其余部分的结构与DDS 相同,仍然具有2 个刚性苯环,因而玻璃化转变温度的下降并不大。从Fig.7(b)不同种类复配固化剂体系的DSC 图中可以看出,20HTDA 体系玻璃化转变温度的降低最大,达到了17.6%,这是因为HTDA 结构中仅有1 个环己基,相较于其他2 种含有苯环的复配固化剂,热性能降低较大;同样摩尔分数 为20% 的20DEDDM 和20DDM 体 系 相 比,20DEDDM 体系的玻璃化转变温度降低较多,这可能是因为DEDDM 两侧苯环上连接的2 个乙基有较大的空间位阻,降低了树脂体系的交联密度,导致玻璃化转变温度降低更多。
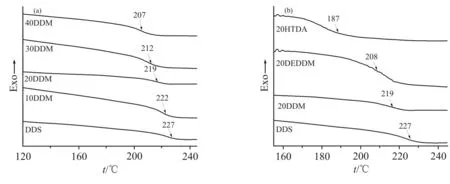
Fig.7 DSC curves of curing productions of different mixed curing agent systems
2.5 不同复配固化剂体系的动态力学性能
对聚合物材料来说,根据其分子链段运动方式的不同,可以分为玻璃态、高弹态和黏流态3 种状态。处于玻璃态的聚合物其分子链段运动被“冻结”,玻璃化转变温度(Tg)是聚合物材料从玻璃态转变为橡胶态的临界温度,在此温度附近,聚合物材料的力学强度和模量等会发生大幅度的降低,通常tanδ值也可代表材料的玻璃化转变温度。Fig.8 为不同复配固化剂树脂体系的动态力学曲线,由图可知,随着DDM 替换量的增加,树脂的tanδ值逐渐降低,且不同种类复配固化剂体系相较于纯DDS 体系tanδ值也有所降低,这与通过DSC 法所测得的结果基本一致。
Fig.9 为不同复配固化剂体系的储能模量-温度曲线。由Fig.9(a)可知,随着DDM 比例的增加,固化产物在50 ℃时的储能模量呈现下降趋势,从4579 MPa 降低至3931 MPa,这是由于DDS 结构中的砜基具有很强的吸电子效应,能很大程度地提高体系的刚性,而复配固化剂体系中,DDM 含量的提高会不断降低DDS 在整个体系中的含量,导致体系刚性下降,储能模量也随之降低。由Fig.9(b)可知,3 种复配固化剂体系相较于纯DDS 体系,储能模量均有所降低,其中HTDA 降低的较多,这是因为HTDA 结构中的环己基相比其余3 种芳香族固化剂中所含有的苯环刚性较低,在相同比例的添加状态下,储能模量降低最大,从4579 MPa 降低至3664 MPa,降低了19.9%。这说明复配固化剂的结构和含量都对环氧树脂固化产物的储能模量有较大影响。
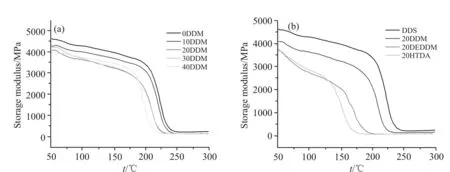
Fig.9 Storage modulus-temperature curves of curing productions of different mixed curing agent systems
2.6 不同固化剂复配环氧树脂体系的热失重分析
Fig.10 为E51/DDS/DDM 体系在氮气气氛下测得的TGA 和DTG 图。热分解温度是评价聚合物耐热性的重要指标,其与聚合物的分子结构有密切联系,凡是能束缚聚合物分子运动的各种因素都与其耐热性相关。在聚合物分子主链上引入芳香环或芳杂环及减少单键,将大大提高分子链的刚性,从而提高其热稳定性。
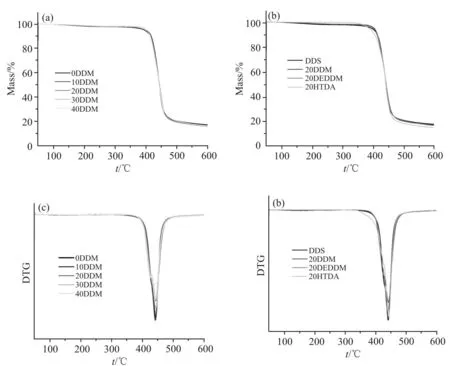
Fig.10 TGA and DTG curves of different mixed curing agents
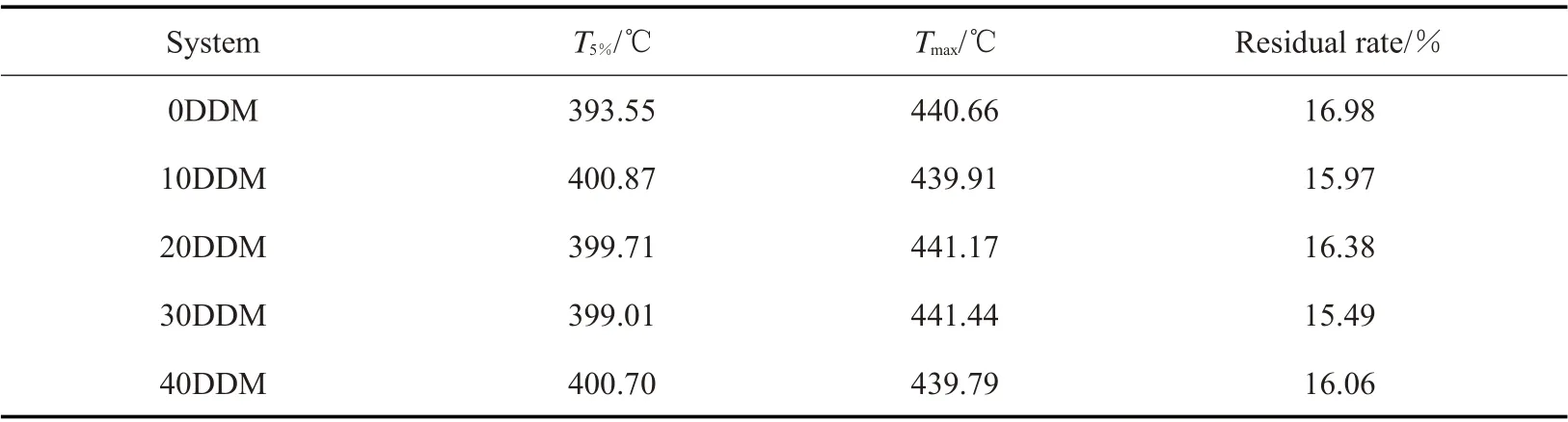
Tab.6 Thermal properties of different contents of mixed curing agent systems
从Fig.10 中可以看出,在替换了一部分DDM后,环氧树脂固化产物的降解均为一步降解,且T5%和Tmax均在400 ℃以上,表现出较强的耐热性能。DDM 的加入对原有体系的热分解温度没有产生很大影响,这主要是因为DDM 和DDS 分子中均含有2个苯环,且其结构相似,胺当量相近,DDM 的加入没有降低原有体系中苯环的含量,使得所得固化产物的热性能没有发生太大变化。
Tab. 7 为相同含量不同种类复配固化剂的TGA和DTG 数据,20DDM 和20DEDDM 的热性能没有发生显著变化,而20HTDA 的T5%从393.55 ℃降低到了385.17 ℃,而最大分解速率则没有发生变化。这是由于HTDA 作为一种脂环族的二胺类固化剂,其取代一部分DDS 后,导致环氧树脂体系中苯环的含量降低,交联体系中含有HTDA 环己基结构的部分在较低温度下分解损失;而DDS 在体系中仍然有着较高的含量,是固化后环氧树脂体系的主要部分,因此最大分解速率没有发生显著变化。
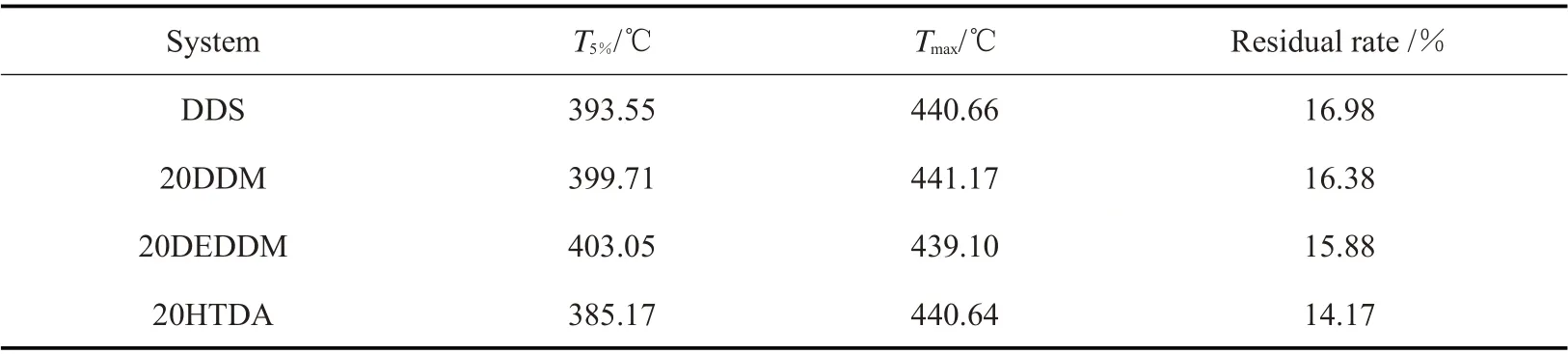
Tab.7 Thermal properties of different kinds of mixed curing agent systems
3 结论
本文以环氧树脂E51 为基体树脂,分别将3 种不同结构的固化剂与芳香胺固化剂DDS 复配,研究了不同固化剂及不同含量的同种固化剂对环氧树脂体系固化动力学及热性能的影响,旨在寻找一种复配固化剂,在降低体系反应活化能和黏度的同时,满足其他性能要求,阐述了复配固化剂结构对性能的影响,旨在为环氧树脂复合材料的制备提供理论依据。
3 种复配固化剂均能有效降低原有体系的黏度和平均反应活化能,复配固化剂的种类和含量对其降低的幅度有较大影响,且复配固化剂的结构对固化产物的性能高度相关。
体系20DDM 与原体系相比,黏度在操作温度80 ℃时降低了70.4%,平均反应活化能降低2.99 kJ/mol,玻璃化转变温度降低至219 ℃,符合耐高温树脂使用要求。综合考虑各方面性能,体系20DDM 可用作环氧树脂复合材料体系的复配固化剂的首选。
猜你喜欢
河北地质(2022年2期)2022-08-22 06:23:54
建材发展导向(2022年12期)2022-08-19 02:31:02
中学化学(2022年5期)2022-06-17 16:51:48
高中数理化(2020年1期)2020-02-29 02:21:18
四川师范大学学报(自然科学版)(2018年2期)2018-04-28 02:21:08
环境科技(2016年1期)2016-11-08 12:17:42
吉林农业(2016年15期)2016-08-30 03:45:48
吉林农业(2016年8期)2016-05-14 13:52:43
工程建设与设计(2016年8期)2016-03-11 15:57:34
郑州大学学报(医学版)(2015年2期)2015-02-27 14:50:53
