
Corrigendum: Purinergic signaling systems across comparative models of spinal cord injury
2022-08-11 06:00
中国神经再生研究(英文版) 2023年3期
Abstract Within the last several decades, the scientific community has made substantial progress in elucidating the complex pathophysiology underlying spinal cord injury. However, despite the many advances using conventional mammalian models, both cellular and axonal regeneration following spinal cord injury have remained out of reach. In this sense, turning to non-mammalian, regenerative species presents a unique opportunity to identify pro-regenerative cues and characterize a spinal cord microenvironment permissive to re-growth. Among the signaling pathways hypothesized to be dysregulated during spinal cord injury is the purinergic signaling system. In addition to its well-known role as energy currency in cells, ATP and its metabolites are small molecule neurotransmitters that mediate many diverse cellular processes within the central nervous system. While our understanding of the roles of the purinergic system following spinal cord injury is limited, this signaling pathway has been implicated in all injury-induced secondary processes, including cellular death, inflammation, reactive gliosis, and neural regeneration. Given that the purinergic system is also evolutionarily conserved between mammalian and non-mammalian species, comparisons of these roles may provide important insights into conditions responsible for recovery success. Here, we compare the secondary processes between key model species and the influence of purinergic signaling in each context. As our understanding of this signaling system and pro-regenerative conditions continues to evolve, so does the potential for the development of novel therapeutic interventions for spinal cord injury.
Key Words: cell death; differentiation; glia; inflammation; neurogenesis; proliferation; purinergic signaling; reactive gliosis; regeneration; spinal cord injury; teleost
Review
Purinergic signaling systems across comparative models of spinal cord injury
Eva E. Stefanova, Angela L. Scott

Introduction 689 Search Strategy and Selection Criteria 690 Cell Death 690 Immune Response 692 Reactive Gliosis 692 Neurogenesis and Axonal Regeneration 693 Concluding Remarks 695
Introduction
Following spinal cord injury (SCI), functional recovery is extremely limited in adult mammals. This lack of recovery is reflective of the collective failure of lesioned axons to re-grow and damaged neurons to regenerate in the spinal cord following the primary trauma. The result is a debilitating reduction in sensorimotor function and quality of life in approximately 770,000 people of all ages, genders, ethnicities, and socioeconomic backgrounds each year (Kumar et al., 2018). In contrast to the devastating consequence of spinal cord injury in mammals, several non-mammalian species within the vertebrate subphylum have a much higher regenerative capacity within the central nervous system (CNS) and can undergo functional recovery in adulthood. Although the underlying source of the regenerative differences between regenerating and non-regenerating species is not fully understood, landmark work studying these comparisons has begun to shed light on the defining features and characteristics of successful regeneration and functional recovery.
While all vertebrates have a certain degree of regenerative capacity in the CNS during development, by adulthood, this plasticity is highly restricted in birds and mammals. Limitations to regeneration are largely attributed to a secondary cellular insult involving acute and chronic processes of neuroinflammation, cellular death, reactive gliosis, and axonal degeneration within the spinal cord following injury. The species who maintain some regenerative ability beyond development appear to be members of the more primitive classes of vertebrates including Agnatha (jawless fish), Reptilia, Amphibia, and Osteichthyes (teleost fish), suggesting that regeneration is an ancestral trait that is diminished over evolution. Indeed, zebrafish (Danio rerio
) (Reimer et al., 2008; Hui et al., 2010; Goldshmit et al., 2012), sea lamprey (Petromyzon marinus
) (Herman et al., 2018), axolotl (Ambystoma mexicanum
) (Sabin et al., 2019), knifefish (Apteronotus leptorhynchus
) (Vitalo et al., 2016), goldfish (Carassius auratus
) (Takeda et al., 2015, 2017), and newts (Notophthalmus viridescens
) (Zukor et al., 2011) are all non-mammalian vertebrate species with a preserved high degree of CNS regeneration capacity. One particularly distinct difference between these select species and mammals is the ability to undergo injury-induced neurogenesis following SCI (Reimer et al., 2008; Takeda et al., 2008; Sirbulescu et al., 2009; Hui et al., 2010; Goldshmit et al., 2012; Joven and Simon, 2018). In adult zebrafish for instance, newly generated motoneurons are integrated into the existing circuitry to restore locomotion below the level of the lesion within 6 weeks post SCI (Reimer et al., 2008). A further understanding of the neuroprotective and regenerative mechanisms in these species will help to uncover the molecular cues associated with limiting secondary injury and promoting regeneration and recovery.One evolutionarily conserved family of signaling factors involved in various aspects of secondary responses to injury in both regenerative and nonregenerative species is the purinergic signaling system. Purinergic signaling has been associated with regulating cellular proliferation, migration, differentiation, and survival during development and in adulthood (Burnstock and Ulrich, 2011; Gomez-Villafuertes, 2016; Oliveira et al., 2016; Ribeiro et al., 2016). Purines and pyrimidines, such as adenosine triphosphate (ATP) and uridine triphosphate (UTP) respectively, along with their metabolites, function as extracellular signaling molecules that mediate cellular communication via purinergic receptors (Burnstock, 2018). These receptors have extensive protein families, including metabotropic adenosine A, A, A, A, ionotropic nucleotide P2X, and metabotropic nucleotide P2Yreceptors (Burnstock, 2018). Following mammalian SCI, Wang et al. (2004) found that extracellular ATP is elevated within the lesion site for several hours, thus suggesting a role for injury-induced purinergic signaling. Since then, it has been proposed that ATP may function as an early distress signal within the tissue that is involved in the chemoattraction of various cell types and induction of cell reactivity and proliferation (Haynes et al., 2006; Suadicani et al., 2006; Dou et al., 2012; Shinozaki et al., 2017; Quintas et al., 2018; Kobayakawa et al., 2019). While accumulating evidence supports a role for purinergic signaling in the CNS injury response of mammals, how this complex system may be adapted in regeneratively competent, non-mammalian species is less clear. Here, we review on the role of purinergic signaling during each phase of the secondary injury [(1) cellular death, (2) neuroinflammation, (3) reactive gliosis, and (4) axonal degeneration] and examine the variations of this signaling system in regenerative and non-regenerative species.
Search Strategy and Selection Criteria
Bibliographic citation databases searched include PubMed and Google Scholar. Search terms include: P1 receptors, P2X receptors, P2Y receptors, teleost spinal cord injury, mammalian spinal cord injury, P1/P2X/P2Y and inflammation, P1/P2X/P2Y and cell death, P1/P2X/P2Y and gliosis, P1/P2X/P2Y and neurogenesis, P1/P2X/P2Y axonal regeneration. All searches were completed between 2020‒2021.
Cell Death
Following an initial mechanical trauma to the spinal cord, secondary injury is associated with a wave of cell death that exacerbates damage and expands the lesion size (Figure 1
). In rodent models of SCI, the predominant mechanism of neuronal cell death is necrosis. This begins almost immediately, peaks at 1‒2 days, and continues for months (Hassannejad et al., 2018; Kwiecien et al., 2020). Glial cells similarly reach peak levels of necrotic cell death between 1‒2 days following SCI (Kwiecien et al., 2020). Meanwhile, apoptosis of neurons peaks first around 4‒8 hours and then again at 3 days (Hassannejad et al., 2018). In oligodendrocytes, it peaks within hours and continues for weeks (Crowe et al., 1997), and in microglia, it peaks at 1 day (Bellver-Landete et al., 2019). Additional mechanisms of secondary cell death include intracellular calcium dysregulation, excitotoxicity, and mitochondrial dysfunction/oxidative stress (Ohishi et al., 2016; Reigada et al., 2017).In contrast, regeneratively competent species predominantly display apoptotic, as opposed to necrotic, cell death following a CNS injury (Figure 2
) (Sirbulescu et al., 2009). Notably, necrosis stimulates a pro-inflammatory immune response that leads to further damage, whereas apoptosis is typically anti-inflammatory (Kono and Rock, 2008). The presence of secondary cell death in regenerative species suggests that some degree of cell death following injury may be tolerated, and perhaps essential, for successful CNS regeneration. Indeed, inhibition of apoptosis following caudal fin amputation in adult zebrafish reduced blastema formation and sensory neuron regrowth (Rampon et al., 2014). Similarly, inhibition of caspase-mediated cell death within the first 24 hours post injury was found to prevent tail regeneration following amputation in the African clawed frog tadpole (Xenopus laevis
) (Tseng et al., 2007). However, the duration of cell death differs significantly between regenerative and non-regenerative species. In teleosts, apoptosis peaks between 15‒24 hours post SCI and then declines sharply after 3‒5 days (Sirbulescu et al., 2009; Hui et al., 2010, 2014). The sustained expression of apoptotic markers observed 3 weeks post SCI in rodents (Crowe et al., 1997) and 8 weeks in humans (Emery et al., 1998) suggests that regenerative failure in mammals may not be caused by the presence of secondary cell death, but by its necrotic and prolonged nature.Activation of purinergic signaling is kicked off very early and known to promote cell death in the damaged mammalian spinal cord (Figure 3
). Apoptosis stimulates damaged cells to release nucleotides into the extracellular space, further stimulating additional ATP release from nearby cells (Elliott et al., 2009; Chekeni et al., 2010). In addition, excess glutamate within the lesion site induces increased Cainflux in adjacent neurons and glia that also stimulates ATP release (Sieger et al., 2012). As a result, an elevated concentration of extracellular ATP in and around the lesion is sustained for several hours following injury (Wang et al., 2004). Local activation of both adenosine and P2X receptors have been implicated in dysregulation of intracellular calcium homeostasis and cell death induction. In particular, activation of both Aand P2Xenables rapid cytosolic Caoverload and subsequent cell death soon after mouse SCI (Wang et al., 2004; Paterniti et al., 2011; Leeson et al., 2018). Moreover, P2Xreceptor activation potentiates P2X-mediated cell death (Kawano et al., 2012b). Purinergic activation of microglia and macrophages within the lesion site also facilitates P2X-mediated cytotoxic cell death of oligodendrocytes (Lopez-Serrano et al., 2019) and apoptosis of many other potential cell types expressing P2X, including spinal neurons, ependymal cells, astrocytes, and inflammatory cells (Wang et al., 2012; Gandelman et al., 2013; Marichal et al., 2016; He et al., 2017; Bidula et al., 2019). Interestingly, P2Xis conserved and expressed ubiquitously within most tissues of zebrafish and other bony fish species (Ogryzko et al., 2014; Rump et al., 2020). However, the exact role of P2Xin mediating injury-induced cell death within the spinal cord of these species has not been explored.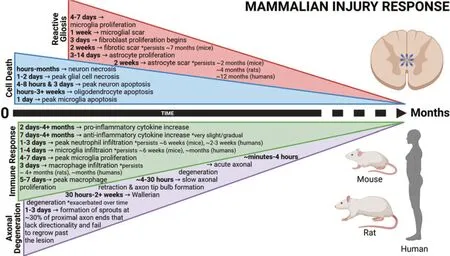
Figure 1 |Timeline depicting the secondary cellular injury response following mammalian spinal cord injury. The primary mechanical trauma is exacerbated by prolonged cell death, widespread inflammation, reactive gliosis, and axonal degeneration. These events prevent successful regeneration and limit sensorimotor recovery. Created with BioRender.com with permissions and publication license.
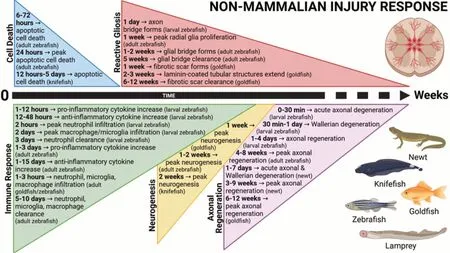
Figure 2 |Timeline depicting the secondary cellular injury response following non-mammalian spinal cord injury. The primary mechanical trauma induces transient cell death, controlled inflammation, reactive gliosis, neurogenesis, and axonal regeneration. These events conclude within weeks and facilitate recovery and restoration of locomotor function in non-mammalian vertebrates. Created with BioRender.com with permissions and publication license.
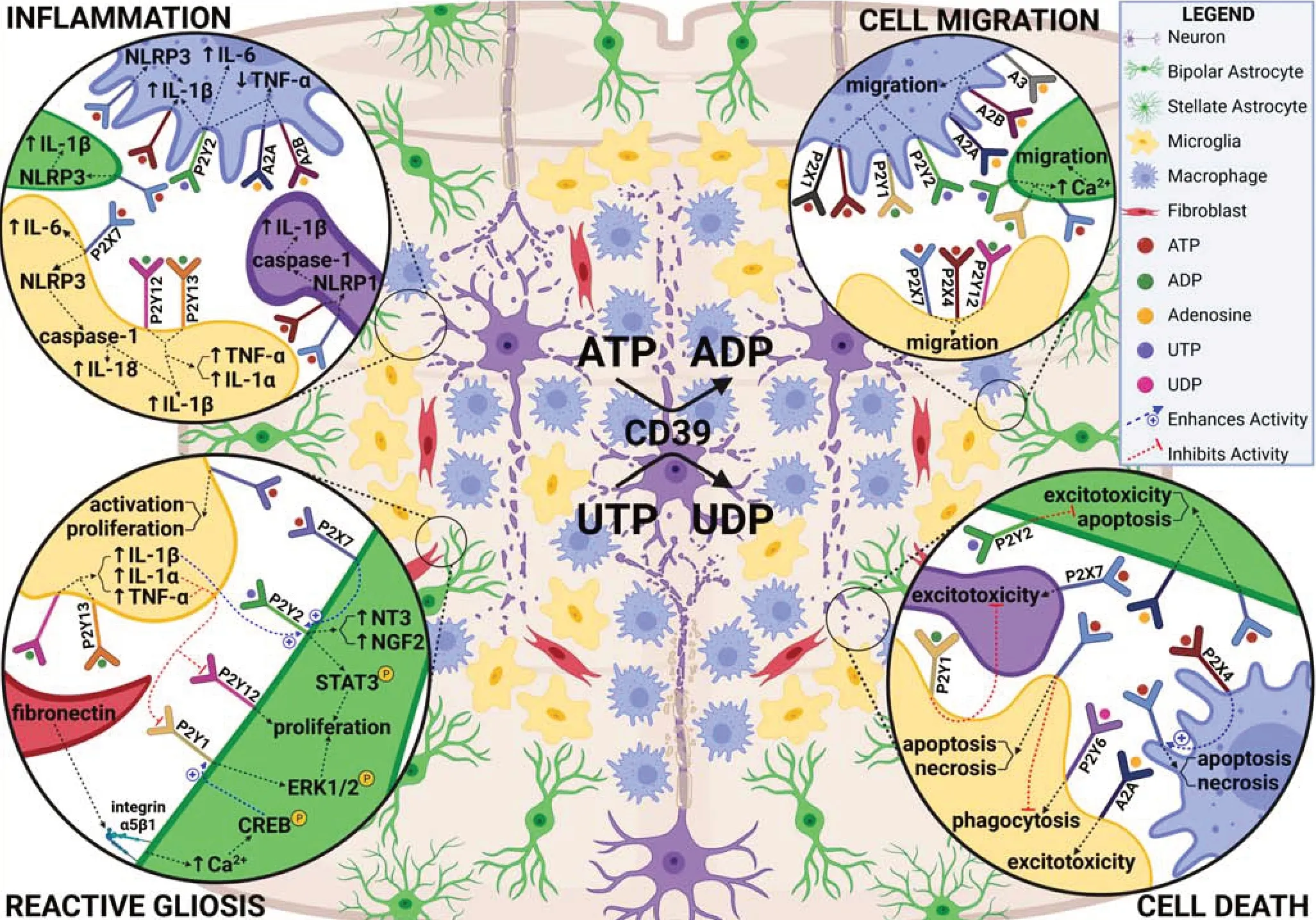
Figure 3 |Purinergic signaling within the spinal cord microenvironment during the early injury response. The first several days following mammalian SCI are characterized by widespread cell death, migration of various cell types to the lesion, inflammation, and reactive gliosis. Identified roles for purinergic receptors in these processes is summarized. Created with BioRender.com with permissions and publication license. ADP: adenosine diphosphate; ATP: adenosine triphosphate; CD39: cluster of differentiation 39; Ca2+: calcium; CREB: cAMP response element-binding protein; ERK1/2: extracellular signal-regulated kinases 1/2; IL: interleukin; NGF2: nerve growth factor-2; NLRP1/3: NLR family pyrin domain containing 1/3; NT3: neurotrophin-3; STAT3: signal transducer and activator of transcription 3; TNF-α: tumor necrosis factor alpha; UDP: uridine diphosphate; UTP: uridine triphosphate. Created with BioRender.com with permissions and publication license.
While purines may exacerbate the initial injury by inducing cell death, there is evidence that they may also induce cell proliferation and neuroprotection. Following caudal fin amputation in zebrafish, activation of Areceptors via adenosine enhanced progenitor cell recruitment, which consequently increased cell proliferation within the lesion beyond spontaneous levels (Rampon et al., 2014). In mammals, microglia P2Yreceptor stimulation enhanced neuronal survival following transient forebrain ischemic injury (Fukumoto et al., 2019), whereas astrocyte and neuronal P2Yreceptor stimulation was associated with the inhibition of apoptosis through activation of ERK1/2 and Akt signaling pathways and upregulation of neurotrophins, growth factors, and anti-apoptotic genes (Chorna et al., 2004; Arthur et al., 2006). Furthermore, pre-treatment of neuroblastoma cells with diadenosine tetraphosphate prior to strong ATP stimulation decreased excitotoxicity (Reigada et al., 2017). This reduced not only P2X, but also P2X, P2Y, and P2Yexpression, probably through activation of P2Yreceptors (Reigada et al., 2017). In the rodent spinal cord, P2Yis widely expressed by neurons, glia, and immune cells and is upregulated from 4‒28 days following injury (Rodriguez-Zayas et al., 2010). Interestingly, stimulation of astrocyte P2Xreceptors induces phosphorylation of ERK1/2 that subsequently increases P2YmRNA expression (D’Alimonte et al., 2007). Whether P2Yand P2Yplay similar roles in regeneratively competent vertebrates following SCI remains unknown.
Immune Response
Shortly after the primary trauma, the microenvironment of the injured mammalian spinal cord is largely occupied by resident and infiltrating inflammatory phagocytic cells (Figure 1
). Microglia, CNS-derived macrophages, undergo cell death within the first 24 hours post injury like other cell types; however, residual microglia also undergo activation and proliferation to subsequently repopulate the spinal cord region (Bellver-Landete et al., 2019). Indeed, rapidly proliferating microglia are the predominant phagocytes of cellular and myelin debris within the first few days after injury (Bellver-Landete et al., 2019). By 7 days, microglia proliferation peaks, and by 14 days, the number of microglia within and around the lesion core reaches maximal levels corresponding to the transformation of the area into a ‘lesion cavity’ (Bellver-Landete et al., 2019; Kwiecien et al., 2020). While few peripheral macrophages and fibroblasts occupy the lesion site at 72 hours post injury, these cells rapidly infiltrate over the course of the following week (Zhu et al., 2015) and soon outnumber microglia (Greenhalgh and David, 2014). The occurrence of phagocytic activity within the spinal cord appears important for recovery, as depletion of spinal cord microglia results in delayed myeloid cell recruitment, increased lesion size, enhanced neuronal and oligodendrocyte cell death, and exacerbated locomotor dysfunction (Bellver-Landete et al., 2019). However, harmful inflammatory cells have been found to persist and phagocytose myelin beyond 4 months post SCI in rats (Kwiecien et al., 2020) and up to one year in humans (Fleming et al., 2006). Thus, the beneficialversus
detrimental effects of inflammatory cells within the spinal cord is largely dependent on their spatial and temporal presence following injury.In comparison to macrophage recruitment in mammals, regeneratively competent teleosts appear to have accelerated recruitment of resident and peripheral immune cells to the injury site (Figure 2
). In goldfish, SCI induces reactive microglia, macrophage, and neutrophil migration to the lesion site within 1 hour post injury, and migration to damaged neurons by 3 hours (Koganti et al., 2020). Similarly, neutrophils, followed by microglia and macrophages, infiltrate the larval zebrafish spinal cord within 2 hours post injury (Tsarouchas et al., 2018). In addition to the advanced infiltration of inflammatory cells, clearance of these cells also occurs at a higher rate in teleost fish. Accumulating macrophages, neutrophils, and monocytes within the spinal lesion of zebrafish are cleared by 5‒10 days post injury (Hui et al., 2010), a much faster process than the prolonged rate in mammals. Indeed, the enhanced immune cell recruitment to, and clearance from, the injury site following a CNS trauma may be one of the key adaptations by this species that distinguishes their regenerative capacity from others.One of the primary signals that draws the migration of immune cells to the lesion site is ATP (Elliott et al., 2009; Kronlage et al., 2010). An ATP gradient, enhanced by ATP-induced-ATP release from local astrocytes (Suadicani et al., 2006), microglia (Dou et al., 2012), and macrophages (Kobayakawa et al., 2019) provides necessary directional cues for their movement towards the injury site within minutes of a CNS trauma (Davalos et al., 2005). In particular, microglial migration in mammals is largely mediated via activation of P2Yand P2Xreceptors (Haynes et al., 2006; Ohsawa et al., 2007); while P2X, P2Y, P2Y, A, A, and Areceptors have been implicated to a lesser degree (Kronlage et al., 2010; Kobayakawa et al., 2019). ATP provides a chemotactic gradient that promotes the extension of pre-existing membrane lamellipodia and activates macrophage membrane propulsion machinery (Kronlage et al., 2010). Inhibition of metabotropic purinergic receptors caused macrophages to scatter throughout the injured cord and enhanced secondary axonal dieback outside the lesion core (Evans et al., 2014). In larval zebrafish, microglial migration is also driven by P2Y(Sieger et al., 2012), whereas macrophage and neutrophil migration is mediated by P2Xreceptors (Ogryzko et al., 2014). The recruitment of peripheral macrophages in larval zebrafish appeared necessary for regeneration as mutants lacking macrophages displayed an extended period of neuroinflammation and impaired axonal regrowth (Tsarouchas et al., 2018).
Once migration to the lesion site occurs, mammalian microglial cells downregulate P2Yand subsequently upregulate P2Xand P2Yreceptors to stimulate proliferation and phagocytotic behavior (Koizumi et al., 2007; Monif et al., 2009; Bellver-Landete et al., 2019). In addition to proliferation, P2Xupregulation also induced the activation of microglia and an increase in pro-inflammatory gene expression (Monif et al., 2009; He et al., 2017). Among the purinergic receptors, P2Xhas been most strongly implicated in mediating pro-inflammatory cytokine expression and secretion by various cell types within the mammalian CNS (Giuliani et al., 2017; He et al., 2017). Resident activated microglia and, to an even greater extent, infiltrating macrophages lead to an early (~2 days post SCI) and late wave (~14 days post SCI) upregulation and release of pro-inflammatory cytokines, including interleukins (IL-1α, IL-1β, IL-6) and tumor necrosis factor-α (TNF-α) (Pineau and Lacroix, 2007; Bellver-Landete et al., 2019; Kwiecien et al., 2020).
In comparison to mammals, SCI in zebrafish induces a brief pro-inflammatory response characterized by elevated expression of IL-1β and TNF-α that also appears to be driven by P2Xactivation (Ogryzko et al., 2014; Tsarouchas et al., 2018). Interestingly, inhibiting the initial pro-inflammatory TNF-α and IL-1β upregulation in larval zebrafish reduced axonogenesis, whereas IL-1β downregulation at later time points was required for successful regeneration (Tsarouchas et al., 2018). The decrease in expression of IL-1β also corresponds with a longer anti-inflammatory response characterized by elevated expression of transforming growth factor- β1a (TGF-β1a) and transforming growth factor-β3 (TGF-β3) by 3 days following injury (Hui et al., 2014; Tsarouchas et al., 2018). This supports the view that an early pro-inflammatory response is adaptive and required for regeneration, but sustained immune responses can be detrimental to recovery.
P2X-mediated effects within the spinal cord appear to be regulated by the purinergic P2Xreceptor. Macrophage P2Xreceptors enhance P2X-mediated pro-inflammatory cytokine release in mice and in this way, help to sustain the neuroinflammatory response following SCI (Kawano et al., 2012a). In contrast, P2XmRNA expression is downregulated with SCI in zebrafish (Hui et al., 2014). While it is unclear what this means in terms of regenerative success, deletion of P2Xdecreases inflammasome activation, IL-1β secretion, and neuronal degeneration, while reducing microglial/macrophage activation, immune cell infiltration, and lesion size following SCI in mice (de Rivero Vaccari et al., 2012). In addition to P2Xand P2X, several other purinergic receptors have also been implicated in driving cytokine release in mammals. These include macrophage P2Yreceptors, which induce an increase in IL-1β, IL-6, and a decrease in TNF-α (de la Rosa et al., 2020), and Areceptors, which similarly decrease TNF-α release (Cohen et al., 2013). Lastly, stimulation of microglial P2Yand P2Yreceptors also appears to induce IL-1β, IL-6, and TNF-α release (Liu et al., 2017). Given some of the key roles of purinergic receptors across various cell types in neuroinflammation (Figure 3
), evidence for a therapeutic advantage by targeting this signaling system during late response phases is growing.Reactive Gliosis
The migration of macrophages and microglia to the lesion core is known to recruit other cell types, including perivascular fibroblasts and astrocytes, that form a multi-layer ‘glial scar’ in mammalian SCI (Figure 1
). Peripheral macrophages appear to be largely responsible for the formation of an inner fibrotic scar around the core of the lesion site that is fully encircled by reactive microglia within the first week following SCI (Zhu et al., 2015; Bellver-Landete et al., 2019). The reactive microglia then work to orchestrate the formation of the outermost portion of the glial scar comprised of newly proliferating GFAPastrocytes (Wanner et al., 2013) (Figure 4
). Depletion of reactive microglia reduces astrocyte proliferation by approximately 40‒55%, which disrupts the organization of the glial scar and further impairs locomotor function (Bellver-Landete et al., 2019). On the flip side, augmenting microglial proliferation during the first week post injury enhances astroglial scar formation, increases cell survival, and improves recovery (Bellver-Landete et al., 2019). This suggests that the astroglial scar component is particularly important for preventing secondary cellular death and promoting functional recovery (Okada et al., 2006; Wanner et al., 2013; Bellver-Landete et al., 2019).The formation of the astroglial scar begins as early as 3 days post SCI, peaks around 7 days, and is fully formed by 14 days (Wanner et al., 2013; Bellver-Landete et al., 2019). During early glial scar formation (5‒7 days), newly proliferated astrocytes become oriented perpendicular to the lesion core and display a bipolar radial-glia-like morphology, unlike their ‘inactivated’ stellate counterparts distal from the lesion (Wanner et al., 2013). Indeed, a gradient spanning the lesion site consists of increasing astroglial proliferation, density, radial-like morphology, and GFAPexpression. During chronic glial scar formation (9‒21 days), reactive bipolar astrocytes transform into scar-forming astrocytes as they form a ‘dense meshwork’ of intersecting and overlapping cell processes that express the inhibitory factors: chondroitin sulfate proteoglycans (CSPGs) (Wanner et al., 2013; Hara et al., 2017). Disrupting early glial scar formation through the inhibition of STAT3 signaling, a critical regulator of astrogliosis, led to a scattering of inflammatory immune cells throughout the injured spinal cord that augmented secondary neuronal and oligodendrocyte cell death (Okada et al., 2006; Wanner et al., 2013). Deletion of the chronic glial scar using diphtheria toxin similarly exacerbated tissue damage (Anderson et al., 2016). However, attenuating the transformation of reactive astrocytes to those that are scar forming during later time frames post injury had a positive effect on axonal regeneration (Hara et al., 2017). Thus, while injury-induced astrogliosis may corral inflammatory cells at the lesion core and promote regeneration early on, it is equally important to limit this process in chronic conditions.
Both purinergic and cytokine-mediated signaling are involved in astrocyte reactivity and proliferation (Figure 3
). How factors of these signaling families contribute to astroglial scar formation appears to be part of a delicate balance of local interactions between astrocytes and microglia. Purinergic signaling via P2Yand P2Yreceptors significantly promotes astrocyte proliferation (Quintas et al., 2018). However, both the presence of microglia and IL-1α/TNF-α prevented P2Yactivation and astrocyte proliferation in response to agonist ADPβS. In the study, authors proposed this was due to the expression of P2Yreceptors by microglia. P2Yis abundantly and exclusively expressed by microgliain vivo
and P2Yantagonism rescues ADPβS mediated astrocyte proliferation in astrocyte/microglia co-cultures (Quintas et al., 2018; Stefani et al., 2018). Thus, stimulation of microglial P2Yreceptors may result in the release of IL-1α and TNF-α, inhibition of astrocyte P2Yreceptors, and prevention of astrocyte proliferation. It is likely that while microglia draw astrocytes to the lesion site, they also act to modulate the degree of astrocyte proliferation in response to elevated local purine levels. Indeed, astrocyte proliferation following SCI peaks within 3‒5 days (Wanner et al., 2013): a point in time when proinflammatory IL-1α and TNF-α expression is also strongly upregulated (Kwiecien et al., 2020).In conjunction with proliferation, cytokine signaling is also involved in regulating astrocyte reactivity and cell survival. Microglia secretion of cytokines IL-1β, IL-6, and TNF-α promotes astrocyte transformation from a basal to a reactive phenotype by the concurrent downregulation of astrocyte P2Yexpression and upregulation of STAT3 phosphorylation (Shinozaki et al., 2017). In this study, P2Yknockdown (KD) in astrocytes promoted astrocyte process extension, GFAPexpression, STAT3 phosphorylation, as well as a smaller injury core, decreased neuronal cell death, and decreased immune cell infiltration (Shinozaki et al., 2017). In contrast, others have demonstrated that astrocyte P2Yupregulation protects against oxidative stress-mediated neuronal damage by inducing increased release of IL-6: a factor that induces astrogliosis, enhances spinal cord progenitor cell proliferation via activation of JAK/STAT3, and promotes neuronal differentiation of neural progenitor cells (Okada et al., 2004; Kang and Kang, 2008; Fujita et al., 2009; Oh et al., 2010). It is likely that the apparent conflicting roles of P2Ystem from the action of additional P2Y receptors also expressed within the glial scar.
In addition to other proinflammatory cytokines, IL-1β expression is also elevated within the first week following SCI in mice (Kwiecien et al., 2020) and treatment of mouse primary striatal astrocytes or mouse primary cortical neurons with exogenous IL-1β increases P2YmRNA expression (Stella et al., 1997; Peterson et al., 2013). Similar to P2Y, P2Yreceptor stimulation has also been shown to induce astrocyte proliferation and STAT3 activation (Chorna et al., 2004; Wu et al., 2018). Thus, P2Ymay work to compensate for P2Yinhibition, particularly in knockout and KD manipulations, and continue to promote astrogliosis independently. Interestingly, treatment with pan P2Y antagonists, which target both P2Yand P2Yreceptors, significantly reduce GFAPexpression within the lesion site and exacerbate secondary cell death (Rodriguez-Zayas et al., 2012), demonstrating the overall neuroprotective role of these factors and the astroglial scar.
While reactive gliosis in regeneratively competent teleosts is reflective of that in mammals, key differences do exist (Figure 2
). Similar to mammals, goldfish and zebrafish generate a fibrotic scar within 1‒3 weeks following a spinal cord hemisection; however, this scar does not impede regeneration as it does in mammals (Takeda et al., 2015; Wehner et al., 2017). Glial cells with elevated GFAPexpression surround the fibrotic scar but do not display a hypertrophied morphology (Goldshmit et al., 2012; Takeda et al., 2015; Herman et al., 2018). In the weeks after injury, laminin-coated tubular structures develop across the fibrous scar, and by 6‒12 weeks, glial processes along with regenerating axons utilize these ‘tunnels’ to cross the injury site (Goldshmit et al., 2012; Takeda et al., 2015). Interestingly, deposition of neural/glial antigen 2, an inhibitory CSPG also upregulated in mammals, is present within the goldfish fibrous scar at 3 weeks post injury but its reduction by 6 weeks aligns with the increase in axon infiltration (Takeda et al., 2015).Fibroblast growth factor (FGF) signaling appears to be particularly important for the formation of the glial bridge. Inhibition of FGF in zebrafish following SCI results in significant reductions to GFAPexpression, as well as a decline in axonal regeneration and functional recovery (Goldshmit et al., 2012). Treatment of primary astrocyte cultures derived from postnatal day 14 marmoset (Callithrix jacchus
) cerebral cortex with human recombinant FGF2 accelerated wound closure in a scratch-wound assay by increasing proliferation, migration, and astrocyte transformation from a stellate/multipolar to an elongated/bipolar morphology (Goldshmit et al., 2012). These findings were replicated in a mouse model of SCI where application of exogenous FGF2 dampened the neuroinflammatory response, decreased levels of CSPGs, and promoted the phenotypic transformation of astrocytes into a bipolar morphology resembling the glial bridge in teleost species (Goldshmit et al., 2014). Interestingly, purinergic signaling governs FGFinduced astrogliosis. On one hand, P2Y activation enhances proliferation of astrocytes induced by FGF2; while P2Xstimulation inhibits proliferation by inducing a state of growth arrest (Neary et al., 2008). In addition to FGF, expression of connective tissue growth factor a (CTGFA) by glial cells accelerates glial bridge formation, enhances axonogenesis, and promotes functional recovery in zebrafish (Mokalled et al., 2016). Purinergicmediated CTGFA secretion via Areceptors also supports a role for purines in promoting glial bridge formation (Chan et al., 2013). Future research is required to improve the current understanding of purinergic influences in mammalian and non-mammalian injury-induced gliosis.Neurogenesis and Axonal Regeneration
In the mid 1990’s, a latent neural stem cell niche lining the rodent central canal was identified (Weiss et al., 1996). It was found that under certain conditions, mammalian spinal ependymal cells give rise to new neurons and gliain vitro
: a feat not normally presentin vivo
. The exception to this is the result of a direct injury, but even then, migration is almost absent, proliferation is minimal, and exclusive glial differentiation accounts for a very small percentage of newly derived astrocytes (Ren et al., 2017). Yet transplantation of spinal ependymal cells into the dentate gyrus of the hippocampus, a neurogenic niche within the adult mammalian brain, induced differentiation of these cells into neurons (Shihabuddin et al., 2000). This emphasized the influence of the spinal cord microenvironment and prompted much work into identifying potential cues responsible for determining the cell fate of endogenous spinal ependymal cells (Becker et al., 2018; Liu and Chen, 2019).Apart from other mammalian species, the existence of injury-induced spinal progenitors in humans has been recently challenged. One study examining the central canal in humans suggested that in contrast to the ependymal layer in rodents, the central canal closes caudal to the brainstem beyond 18 years of age (Garcia-Ovejero et al., 2015). The authors show that the ependymal layer is replaced by a dense accumulation of GFAPastroglial processes, ependymocytes, and perivascular pseudorosettes. Furthermore, expression of stem cell markers is highly limited within this region (Garcia-Ovejero et al., 2015). In conjunction, a post-mortem study of human SCI patients (aged 33‒88) found no evidence for injury-induced proliferation of cells within the ependymal region (Paniagua-Torija et al., 2018). Together, these studies suggest that morphological and molecular profiles in adult humans may be quite unique compared to other vertebrate species, and approaches to induce endogenous neurogenesis could be more applicable to both younger individuals and those with higher level injuries.
Another regenerative strategy to encourage functional recovery following SCI is the re-growth of damaged axons or compensatory axonal sprouting. Unfortunately, axonal regeneration and sprouting are also highly restricted in mammalian species (Figure 1
). Within 30 minutes following injury, neurons undergo rapid and symmetrical proximal and distal axon end fragmentation, which is followed by a period of slow axonal retraction until ~30 hours post transection (Kerschensteiner et al., 2005). After this point the injury extends beyond the proximal axon ends. The distal axon ends begin to undergo Wallerian degeneration mediated by infiltrating peripheral macrophages for several days to weeks (Evans et al., 2014). Although limiting macrophage migration to the injury site does not promote axonal survival, restricting them to the injury site does promote distal axonal sparing (Kobayakawa et al., 2019). Interestingly, approximately 30% of transected axons do undergo compensatory sprouting at their proximal ends (Kerschensteiner et al., 2005). While these sprouts grow relatively rapidly and straight within the first two days, they quickly lose directionality and are unable to grow across the injury site. This is largely due to the presence of numerous growth inhibitory environmental cues at the injury site that lead to dystrophic growth cone formation and prevent axonal regeneration (Filous and Schwab, 2018).In contrast to mammals, adult teleost fish and urodele amphibians achieve functional recovery by not only successfully replacing lost neurons, but also regenerating damaged axons (Figure 1
). Radial glia (RG), referred to as ependymoradial glia, are neural stem cells within the CNS of nonmammalian vertebrates that serve many homeostatic roles and are essential for regeneration in zebrafish (Reimer et al., 2008; Hui et al., 2010; Briona and Dorsky, 2014), goldfish (Takeda et al., 2008), brown ghost knifefish (Zupanc, 2019), and salamanders (Joven and Simon, 2018). Morphologically, they line the central canal and extend radial processes out to the pial surface. Functionally, they resemble mammalian astrocytes and ependymal cells (Becker et al., 2018). Following SCI, RG undergo a massive proliferative response reaching peak values at 7 days before differentiating into both neurons and glial cells that later populate the injured cord (Hui et al., 2015). In addition, a subpopulation of RG also appears to contribute to the formation of the astroglial bridge that promotes and guides axonal regeneration in these species (Goldshmit et al., 2012). RG cells are present in the mammalian spinal cord during development and give rise to new neurons, macroglia populations, and ependymal cells (Malatesta et al., 2000; Barry and McDermott, 2005; Xing et al., 2018). However, they undergo terminal differentiation into astrocytes during early postnatal development and are relatively absent from the adult spinal cord (Barry and McDermott, 2005).The regeneration of axons across the lesion site in these non-mammalian species occurs simultaneously with injury-induced neurogenesis. In larval zebrafish, acute axonal degeneration occurs within the first 30 minutes following Mauthner cell axotomy, which is followed by additional retraction and Wallerian degeneration that concludes by 24 hours (Hu et al., 2018). Remarkably, this study showed that by 96 hours post axotomy, axonal regeneration across the lesion reached 78% of the uninjured axon length. More in line with axonal regeneration in mammals, spinal cord transection in newts results in axonal retraction and dystrophic growth cone formation within the first week post injury (Zukor et al., 2011). However, by 2 weeks, growth cones form at some axon tips and ‘wisping axons’ start to grow into the injury site in association with regenerating meninges and RG cell extensions that innervate their targets by 9 weeks (Zukor et al., 2011). Thus, the response to axonal transection in these species appears to be like mammals during early time points (AAD, retraction, Wallerian degeneration, sprouting), but differs significantly during more chronic responses (Figure 2
). Identifying the factors and conditions present in regeneratively competent species, particularly at the later time points; therefore, may be of most use in promoting axonal regeneration in mammals.
Figure 4 |Purinergic signaling within the spinal cord microenvironment during the chronic injury response. After the first week following mammalian SCI, reactive astrocytes become scar forming, ependymal and neural progenitor cells fail to undergo injury-induced proliferation and neuronal differentiation, and axons continue to degenerate. Identified and hypothesized roles for purinergic receptors in these processes is summarized. Created with BioRender.com with publication permissions and publication license. ADP: Adenosine diphosphate; Akt: protein kinase B; ATP: adenosine triphosphate; BDNF: brain derived neurotrophic factor; CD39: cluster of differentiation 39; Ca2+: calcium; CREB: cAMP response element-binding protein; ERK1/2: extracellular signal-regulated kinases 1/2; IL: interleukin; NGF2: nerve growth factor-2; NLRP1/3: NLR family pyrin domain containing 1/3; NT3: neurotrophin-3; STAT3: signal transducer and activator of transcription 3; TNF-α: tumor necrosis factor alpha; UDP: uridine diphosphate; UTP: uridine triphosphate; Wnt: wingless-related intergration site. Created with BioRender.com with permissions and publication license.
Regardless of their ability to achieve functional recovery, non-mammalian vertebrate CNS regeneration does not result in a return to uninjured conditions. First, newly generated interneurons in larval zebrafish appear morphologically and functionally distinct from their uninjured counterparts following SCI (Vasudevan et al., 2021). In addition to having a smaller soma, they have a higher input resistance, a more depolarized resting membrane potential, lower firing frequencies, and lower spike frequencies; however, they are sufficient to restore function. Second, only a subset of neurons undergo axonal regeneration in non-mammalian species studied to date, including newts (Zukor et al., 2011), goldfish (Takeda et al., 2015), and zebrafish (Goldshmit et al., 2012). Finally, while some regenerating axons travel along their original trajectories, as observed in newts (Zukor et al., 2011) and larval zebrafish (Hu et al., 2018), many take novel routes as seen in adult zebrafish (Becker and Becker, 2001). Notably, regenerative ability appears to decline with age in both mammals (Geoffroy et al., 2016) and non-mammalian vertebrates (Edelmann et al., 2013). Further examination of the post injury cytoarchitecture, the regulatory cues present in the microenvironment, and the changes to both over time are all important considerations in understanding regeneration in these species.
The role of purinergic signaling in injury-induced neurogenesis and axonal regeneration is quite limited (Figure 4
). Expression of several purinergic receptors have been identified in mammalian spinal ependymal cells, including P2Y, P2Y, P2X, and P2X(Gomez-Villafuertes et al., 2015). In response to SCI, the expression of P2Yreceptors is downregulated, whereas expression of P2Y, P2X, and P2Xreceptors is upregulated (Gomez-Villafuertes et al., 2015). How the differential expression of these subtypes in ependymal cells influences their response to injury is largely unknown, but has been shown to play roles in both proliferation and differentiation of other cell types. For instance, P2Yreceptor activation induces proliferation of astrocytes, RG, and tanycytes within the ependymal cell lining of the third ventricle (Weissman et al., 2004; Quintas et al., 2018; Recabal et al., 2021). Similarly, P2Xreceptor expression has been associated with enhancing growth factor expression (e.g. brain-derived neurotrophic factor) and indirectly promoting ependymal cell proliferation (Xu et al., 2018; Ma et al., 2019; Su et al., 2019). In terms of differentiation, P2Yexpression accompanies glutamatergic differentiation of embryonic stem cells, and P2Xstimulation regulates neuronal differentiation of both embryonic and adult neural progenitor cells (Tsao et al., 2013; Glaser et al., 2014; Uda et al., 2016; Leeson et al., 2018). It is likely that the balance and timing of purinergic receptor expression influences the response of ependymal cells to SCI, but further work is needed to determine their roles.In addition to those mentioned above, purinergic receptors P2Y, P2Y, and Ahave also been shown to regulate progenitor cell proliferation, prevent secondary injury, or promote functional recovery. In a recent study of P2Yknockout mice, there was a significant reduction in the number of proliferating neural progenitor cells within the subgranular zone of the dentate gyrus of the hippocampus and the subventricular zone of the lateral ventricles, demonstrating that P2Yreceptor expression is important for progenitor cell proliferation within the adult mammalian brain (Ali et al., 2021). Meanwhile, P2Yknockout mice displayed enhanced progenitor cell proliferation within the dentate gyrus of the hippocampus in early and late adulthood, as well as an increase in neurogenesis (Stefani et al., 2018). Interestingly, inhibition of vesicular nucleotide transporter in postnatal cerebellar cells inhibited quiescence and self-renewal to drive cell-cycle exit toward a neurogenic fate (Paniagua-Herranz et al., 2020). Mechanistically, vesicular nucleotide transporter is responsible for promoting vesicular ATP storage; therefore, inhibition of this transporter decreases ATP signalling and suggests that variation in purinergic ligand alone is sufficient to influence neurogenesis (Paniagua-Herranz et al., 2020). Following SCI, activation of the Areceptor increases Wnt3a and β-catenin mRNA expression in mammals resulting in reductions of inflammation and neuronal cell death (Irrera et al., 2018). While a general reduction in Wnt/β-catenin signaling is typically observed with mammalian SCI (Gonzalez-Fernandez et al., 2014; Irrera et al., 2018), this pathway appears upregulated following SCI in zebrafish and in sea lamprey (Briona et al., 2015; Strand et al., 2016; Herman et al., 2018). Wnt/β-catenin specifically promotes and appears necessary for neuronal differentiation of RG following SCI in larval and adult zebrafish (Briona et al., 2015; Strand et al., 2016). Various interventions targeting the upregulation of canonical Wnt/β-catenin signaling, including Aagonism, following mammalian CNS injury have been widely successful in limiting neuroinflammation and secondary cell death, and enhancing functional recovery (Shruster et al., 2012; Irrera et al., 2018; Xu et al., 2019).
Concluding Remarks
Regenerative failure following injury to the mammalian CNS is the result of a microenvironment that enhances neuroinflammation, pathological reactive gliosis, and neurodegeneration. Studying regeneratively competent species provides an opportunity to elucidate critical pro-regenerative pathways. Given the evolutionary conservation and prevailing role of purinergic signaling in many of these processes throughout development and adulthood, identification of purinergic influences on regenerative processes may be integral to understanding differences among regeneratively competent and non-competent vertebrates. Ultimately, it is the hope that understanding these differences will lead to therapeutic approaches that target both the endogenous progenitor potential of spinal ependymal cells and functional axonal regeneration following spinal cord injury.
Author contributions:
Both authors participated in the preparation of the manuscript and approved the final manuscript.
Conflicts of interest:
The authors declare no conflicts of interest.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:
Rosa Gomez-Villafuertes, Universidad Complutense de Madrid, Spain.

- 中国神经再生研究(英文版)的其它文章
- Inflammation and retinal degenerative diseases
- Synaptic alterations as a common phase in neurological and neurodevelopmental diseases: JNK is a key mediator in synaptic changes
- Brain-derived neurotrophic factor in main neurodegenerative diseases
- The best of both worlds: mastering nerve regeneration combining biological and nanotechnological tools
- Exosomal miR-23b from bone marrow mesenchymal stem cells alleviates oxidative stress and pyroptosis after intracerebral hemorrhage
- Chlorogenic acid alleviates hypoxic-ischemic brain injury in neonatal mice