
Synaptic alterations as a common phase in neurological and neurodevelopmental diseases: JNK is a key mediator in synaptic changes
2022-08-11 06:00ClaraAliceMusiCarloBonadonnaTizianaBorsello
中国神经再生研究(英文版) 2023年3期
Clara Alice Musi, Carlo Bonadonna, Tiziana Borsello
Brain synapses play a key role in neuronal communication: this “conversation” is at the basis of all brain activities and synaptic dysfunction leads to brain disorders. We study the modulators of this crucial synaptic function and we here present the evidence supporting the c-Jun N-terminal kinase (JNK) pathway as a pivotal actor in this scenario.
The term “synapse” is derived from the Greekσύν
(“together”) andἅπτειν
(“tap”, “connect”) meaning “conjunction”. Synapses are highly specialized cellular connections that represent the basic structural units of neuronal communication. There are two different types of synapses: in electrical synapses, presynaptic and postsynaptic cell membranes are connected by special junctions called gap junctions, channels that allow ions to pass from one neuron to another. In chemical synapses, representing the majority of synapses in the mammalian nervous system, the electrical activity in the presynaptic neuron is converted into the release of a chemical neurotransmitter that binds to receptors located in the plasma membrane of the postsynaptic cell. Therefore, two separate cellular components build up the chemical synapse: the pre-synaptic element, which specialized in the release of the neurotransmitter, and the post-synaptic element, which binds the chemical compounds converting them into an electrical signal. The pre- and postsynaptic elements are associated and mirror each other changes, so that changes in one element will correspond to a variation in the second.What is described above identifies the old “bipartite” synapse, more recently updated with the addition of a third structural element: tiny astrocytic processes. This astrocytic component responds to synaptic activity and, in turn, participates in the regulation of synaptic transmission. Nowadays we also consider a fourth cell type, the microglia that makes brief, repetitive contacts with synapses. These dynamic interactions take place in health but also in pathological conditions, like Alzheimer’s disease (AD), where microglia and immune molecules participate in eliminating the dysfunctional synapses (Wilton et al., 2019).
Synapses, both excitatory and inhibitory, are fundamental in shaping brain function and undergo dynamic changes. The dynamic changes of synapses are mainly studied by the visualization of the post-synaptic structure called spines. In fact, by following spines in the living brain it was discovered that they were kept in a state of equilibrium guaranteed by their continuous formation and elimination, resulting in decreased or increased spine turnover, or temporarily shifting to facilitate their additions or eliminations (Holtmaat et al., 2005).
It is therefore very important to keep in mind that a single spine is in a constant evolution; it changes its shape according to the needs and activity of the pre- and post-synaptic neurons and of the neuronal network. This phenomenon is known as synaptic plasticity, a very important feature of both inhibitory and excitatory neurons, by which they can adapt themself to environmental changes. Inhibitory synapses are difficult to visualize and are less studied, thus resulting in biology modifications poorly understood compared to the excitatory synapses. However, inhibitory synapses are fourfold more dynamic than their shaft counterparts (Chen et al., 2012).
The plasticity of the brain is fundamental to preserving its adaptive functionality at the basis of learning and memory, but also stress stimuli can induce structural changes at the synapse level. Growing evidence suggests that, in brain diseases, the first neurodegenerative mechanism takes place at the synapse. It is, therefore, crucial to understand the intracellular mechanisms underlying synaptic modulation to design powerful neuroprotective strategies.
With this in mind, we decided to examine synaptic dysfunction as a common alteration o f n e u ro b i o l o g i c a l m e c h a n i s m s f ro m neurodevelopmental (Angelman and Rett syndromes) to chronic neurodegenerative diseases (Alzheimer and Tauopathy).
Plasticity is a central feature in the developing as well as in the adult brain. Several evidence show that neurodevelopmental disorders, especially Autism spectrum Disorders (among them Angelman, Rett and Dravet syndromes, X fragile, etc.), are characterized by too many synaptic connections causing hyper connectivity of brain circuits. On the contrary, studies on aging and neurodegenerative brain diseases revealed an opposite trend with loss of synaptic connection (Penzes et al., 2011).
In this context, synaptic dysfunction, which usually precedes neuronal death, has been linked to many neurodegenerative diseases, such as Parkinson’s disease, Huntington’s disease and AD, as well as neuropsychiatric diseases. Many mutations in the human synapse proteome (the “synaptome”) have been described to underlie psychiatric and neurological disorders.
Synaptic dysfunction is an emerging hypothesis explaining affective disorders and it was shown that loss of synaptic homeostasis in specific networks also contributes to chronic pain (Torres et al., 2017). In addition, an imbalance at the network level between excitatory and inhibitory synaptic signals gives rise to epilepsy and such an imbalance has also been demonstrated in neurodevelopmental disorders such as Rett and Angelman syndromes and in other autism spectrum disorders (Canitano and Palumbi, 2021).
Although brain disorders present many different pathological/clinical manifestations, as well as causes (i.e. brain ischemia: disruption of blood flow; Parkinson’s disease: degeneration of the dopaminergic neurons in the substantia nigra) transduced by different intracellular molecular pathways, they all have synaptopathy in common. A better understanding of synaptic dysfunction mechanisms will therefore pave the way to new therapeutic strategies exploitable in many different brain diseases thus promoting neuroprotection against neurodegenerative/neurodevelopmental aberrant mechanisms.
Thus, the central focus of our research is to study the key proteins in synaptic dysfunction/dysmorphogenesis. These first synaptic changes represent a promising therapeutical strategy and a crucial temporal window for neuroprotective treatment. In fact, synapses dynamically change and react to stress stimuli passing through an initial reversible phase, during which synaptic function is impaired but can be rescued, to a second phase, if stress persists, where synaptic injury becomes irreversible, and progresses to synaptic death/loss, and eventually neuronal death.
Here were portedour studieson neurodevelopmental (Angelman and Rett syndromes) as well as neurodegenerative diseases (Alzheimer and Tauopathy) (Sclip et al., 2014; Buccarello et al., 2018; Musi et al., 2020, 2021). Previous evidence from our work showed that JNK signaling pathway is involved in stress response as well as in other different physiological functions, and it is a common actor in the synaptopathy of all these brain diseases.
Why JNK is a pivotal key player in neurodegenerative mechanisms? Because JNK modulates 3 major actions in neurons that are highly polarized cells. Indeed, JNK plays diverse functions in different cellular compartments: 1 - at the pre-synaptic terminal, it regulates the zipping of the vesicles and the release of neurotransmitters (Biggi et al., 2017); 2 -at the post-synaptic level, it controls the correct organization of receptors and scaffold proteins in the active zone of the post-synaptic density region (PSD) where the reception of the neurotransmitter (Sclip et al., 2014) takes place; 3 -in the soma, it governs apoptotic, necrotic and autophagic neuronal deaths, acting on different targets and intracellular organelles (Figure 1
). More in detail, in the presynaptic terminal JNK interacts with the t-snare proteins, phosphorylating Syntaxin-1, Syntaxin-2, and Snap25-JNK, influencing the SNARE complex formation of the synaptic vesicles and modulating the zipping and release of the vesicles. On the other hand, in the post-synaptic terminal JNK interacts and phosphorylates the most abundant scaffold protein of the PSD region PSD-95 and also Shank3, these scaffolds regulate the N-methyl-D-aspartatic acid and aminomethylphosphonic acid receptors in the PSD region (Kunde et al., 2013; Musi et al., 2022). In the soma JNK interacts with transcriptional factors, regulating c-Jun, activating transcription factors 2, and c-Fos in the nucleus, as well as with mitochondrial (among them Bcl2, Bclx, Beclin 1) and Golgi (repressing secretory trafficking) machinery. JNK has many somatic targets like caspases, Neurofilament H, SCG10, BAD, BIM family, and many others. JNK is also implicated in axonal transport(de Los Reyes Corrales et al., 2021), another important function in neurons. All these physiological mechanisms are vital in a healthy brain but can be compromised leading to brain disorders.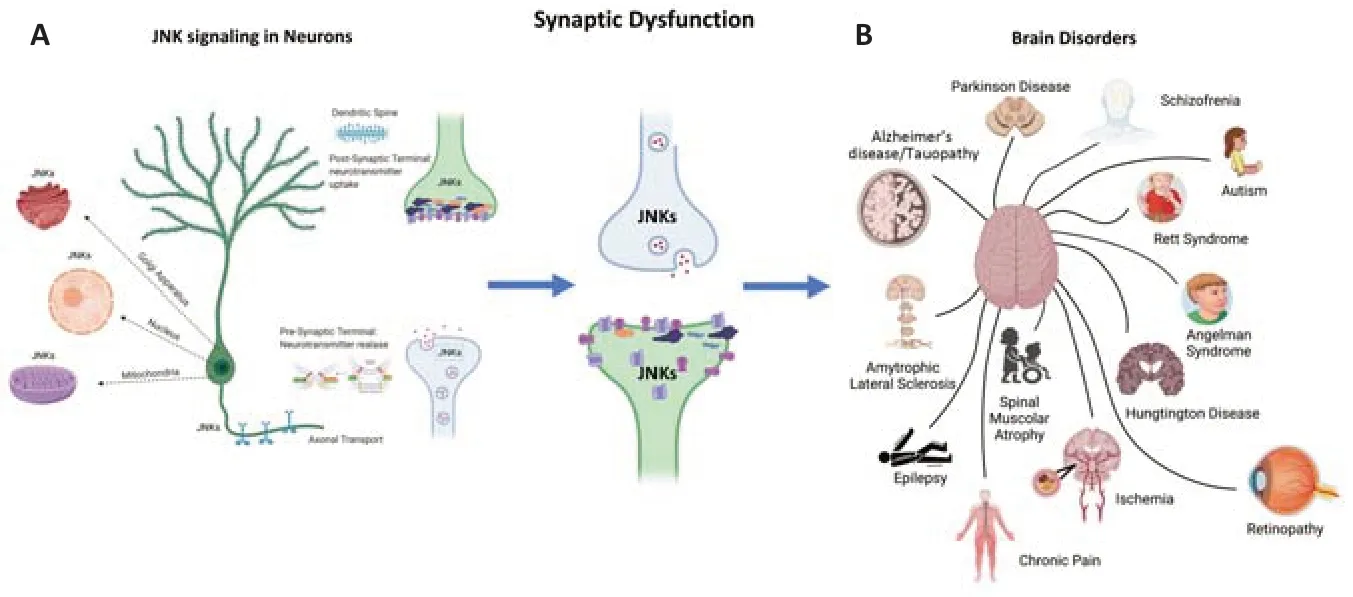
Figure 1 |Brain disorders are characterized by a shared feature called synaptic dysfunction (A) and JNKs localizations in neurons (B). Both neurodegenerative and neurodevelopmental disorders are characterized by synaptic dysfunction. This is the earliest and most common cellular event in all these diseases. The JNK pathway is a key modulator of synaptic dysfunction. There are several pools of JNKs in different cell compartments. These JNKs pools act on different targets. Here we represent different JNK pools (axonal, mitochondrial, nuclear, Golgi, pre-synaptic and post-synaptic pools). The JNK’s targets in these diverse compartments have different functions that influence both the physiology and pathology of neurons. Created with BioRender.com.
In this perspective, we examined JNK activation in Rett and Angelman Syndromes and in AD and tested whether its specific inhibition would provide neuronal protection. We selected two developmental disorders and a neurodegenerative disease (AD), as we are searching for basic cellular and intracellular mechanisms and common key players that govern synaptic dysfunction.
More in detail, in the context of the Rett Syndrome, a rare severe developmental disease, characterized by pseudo-normal development, in which patients usually achieve normal neurodevelopmental milestones but start to regress between 8 and 36 months of age with loss of language and fine motor coordination, locomotive impairment and hand stereotypes, previous work of our group investigated the synaptic dysfunction in two different mouse models (Musi et al., 2021). We identified JNK as an important actor downstream of methyl CpG binding protein 2 (MECP2), the gene mutated in the pathology. Both mice models displayed molecular disorganization of the PSD region in the post-synaptic element. Its disorganization has been related to locomotor and cognitive impairments that characterized this disease. We demonstrated that the specific inhibition of JNK by D-JNKI1, a cell-permeable peptide, strongly ameliorates the symptoms and the disorganization of the PSD region in both mice models. To understand the molecular mechanisms involved in human disease, we used a model of patient induced pluripotent stem cell (iPSC) derived from fibroblast of Rett patients. We demonstrated JNK activation also in human neurons, differentiated from MECP2-mutated iPSCs, compared to the isogenic control expressing wild-type MECP2 allele. The JNK signal was activated in the MECP2-mutated iPSCs and not in control iPSCs, and D-JNKI1 blocked the MECP2mut -induced neuronal death (Musi et al., 2021). Importantly, this is the first proof of concept that JNK is a key player in Human Rett syndrome.
In Angelman Syndrome, another genetic developmental disease characterized by autistic feature, mental retardation, and locomotor disabilities, we analyzed the synaptic dysfunction in the UBE3Amouse model, being UB3A the gene mutated in this syndrome (Musi et al., 2020). JNK was strongly activated also in the brain of these mice, suggesting its important role also in this neurodevelopmental disorder. In line, these mice displayed deregulation of the markers for the excitatory spines. D-JNKI1 treatment improved their behavioral defects, and this correlated with the stabilization of the synaptic biomarkers (Musi et al., 2020).
In neurodegenerative diseases, we mainly studied AD, since JNK phosphorylates both amyloid precursor protein and Tau, the two key proteins of AD, accelerating the formation of β-amyloid oligomers and the deposition of neurofibrillary tangles. JNK regulates the synaptic dysfunction process inin vivo
but alsoin vitro
Alzheimer’s disease models. Indeed, in AD animal model JNK inhibition strongly improved the synaptic impairment also ameliorating the cognitive performances (Sclip et al., 2014).To summarize, the data obtained in these studies strongly support the notion that JNK is a central actor in the degeneration mechanisms of the synapses that characterize both neurodevelopmental and neurodegenerative diseases (Figure 1
).However, JNKs are a family (Jnk1, Jnk2, and Jnk3) of mitogen-activated protein-kinases in which, JNK1 and 2 are ubiquitous, while JNK3 is expressed mainly in the neuronal tissue and is the most highly responsive isoform to stress in the brain pathological context, thus representing a more intriguing and specific target. JNK3 displays an important role in a mouse model of AD (5×FAD mice) and, in line, increased levels and activation of JNK3 have been found in the postmortem brain of AD patients but also in their cerebrospinal fluid (Gourmaud et al., 2015). These indications corroborate the idea that JNK3 is a new therapeutical target to tackle AD and other brain diseases, strongly governed by synaptic dysfunction.
In the past, kinase inhibitors were considered dangerous, with non-specific activities and therefore very difficult to apply in clinical studies. However, thanks to the discovery of specific protein-protein interactions to specifically inhibit kinases, the field is growing. Nowadays, in the field of drug discovery, kinases have become one of the most important targets in chronic and acute diseases. In fact, currently, there are 68 FDAapproved drugs targeting different protein kinases, six of which were approved over the last year.
It is of note that the specific JNK3 inhibition may be used to modulate synaptic changes and prevent synaptic dysfunction in many different brain diseases, as shown by our research and other group’s work. As brain damage, underlying synaptic dysfunction still represents the highest burden for society, we strongly believe that is crucial to develop a strategy to protect synapses. We are now testing JNK3 inhibitors in new neurodegenerative models focusing on neuroinflammation to understand the potentiality of this specific inhibition to tackle central nervous system dysfunctions.
We thank Prof. E. Welker for his advice and criticisms and Prof. M. Repici to revise the manuscript. We acknowledge support from the University of Milan through the APC initiative. The grant from MIUR Project Excellence and the UNIMI Research Support Plan, line 2 action C.
This work was funded by Ricerca Finalizzata
2016 RF-2016-02361941, MIUR, -PON “Ricerca e Innovazione” PerMedNet id project ARS01_01226-PROGETTI DI RICERCA DI RILEVANTE INTERESSE NAZIONALE Prot. 2017MYJ5TH and 312 European Commission’s Horizon 2020 research and innovation program, No. 847749.
Clara Alice Musi, Carlo Bonadonna, Tiziana Borsello
Department of Pharmacological and Biomolecular Sciences, Università degli Studi di Milano, Milano, Italy (Musi CA, Bonadonna C, Borsello T)Mario Negri Insitute for Pharmacolgical Research ‒ IRCCS, Milano, Italy (Musi CA, Borsello T)
Correspondence to:
Tiziana Borsello, PhD, tiziana.borsello@unimi.ithttps://orcid.org/0000-0002-9729-7642 (Tiziana Borsello)
Date of submission:
March 7, 2022Date of decision:
May 7, 2022Date of acceptance:
May 18, 2022Date of web publication:
June 2, 2022https://doi.org/10.4103/1673-5374.346488
How to cite this article:
Musi CA, Bonadonna C, Borsello T (2023) Synaptic alterations as a common phase in neurological and neurodevelopmental diseases: JNK is a key mediator in synaptic changes. Neural Regen Res 18(3):531-532.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Inflammation and retinal degenerative diseases
- Brain-derived neurotrophic factor in main neurodegenerative diseases
- The best of both worlds: mastering nerve regeneration combining biological and nanotechnological tools
- Exosomal miR-23b from bone marrow mesenchymal stem cells alleviates oxidative stress and pyroptosis after intracerebral hemorrhage
- Chlorogenic acid alleviates hypoxic-ischemic brain injury in neonatal mice
- DUSP2 deletion with CRISPR/Cas9 promotes Mauthner cell axonal regeneration at the early stage of zebrafish