
DUSP2 deletion with CRISPR/Cas9 promotes Mauthner cell axonal regeneration at the early stage of zebrafish
2022-08-11 06:00GuoJianShaoXinLiangWangMeiLiWeiDaLongRenBingHu
中国神经再生研究(英文版) 2023年3期
Guo-Jian Shao , Xin-Liang Wang , Mei-Li Wei, Da-Long Ren, , Bing Hu
Abstract Axon regeneration of central neurons is a complex process that is tightly regulated by multiple extrinsic and intrinsic factors. The expression levels of distinct genes are changed after central neural system (CNS) injury and affect axon regeneration. A previous study identified dusp2 as an upregulated gene in zebrafish with spinal cord injury. Here, we found that dual specificity phosphatase 2 (DUSP2) is a negative regulator of axon regeneration of the Mauthner cell (M-cell). DUSP2 is a phosphatase that mediates the dephosphorylation of JNK. In this study, we knocked out dusp2 by CRISPR/Cas9 and found that M-cell axons of dusp2‒/‒ zebrafish had a better regeneration at the early stage after birth (within 8 days after birth), while those of dusp2+/‒ zebrafish did not. Overexpression of DUSP2 in Tg (Tol 056) zebrafish by single-cell electroporation retarded the regeneration of M-cell axons. Western blotting results showed that DUSP2 knockout slightly increased the levels of phosphorylated JNK. These findings suggest that knocking out DUSP2 promoted the regeneration of zebrafish M-cell axons, possibly through enhancing JNK phosphorylation.
Key Words: axon regeneration; central nervous system; CRISPR/Cas9; DUSP2; JNK; Mauthner cell; single-cell electroporation; spinal cord injury; two-photon axotomy; zebrafish
Introduction 577 Methods 578 Results 578 Discussion 580
Introduction
After central nervous system (CNS) injury, the regeneration of axon structure and the reconstruction of synaptic connections are crucial for the recovery of CNS function. While the peripheral nerves of mammals and the CNS of lower model animals, such as zebrafish, have a strong regeneration capacity (Becker and Becker, 2015; Ohnmacht et al., 2016), the CNS in mammals has limited capacity to complete morphological regeneration and functional recovery after injury. Multiple extrinsic and intrinsic factors regulating axonal regeneration have been identified from research on axonal regeneration of various neural systems. Extrinsic factors include acute inflammation (Ohnmacht et al., 2016; Tsarouchas et al., 2018; Wan et al., 2021) and oligodendrocyte myelination (Tsata et al., 2020). Intrinsic factors include intracellular calcium mobilization (Chen et al., 2019) and c-Jun N-terminal kinase (JNK) signaling pathways (Bremer et al., 2019; Chen and Li, 2022).
Spinal cord injury of zebrafish causes changes in the expression level of various genes (Mokalled et al., 2016). We analyzed high-throughput sequencing data and found dusp2 as an upregulated gene in zebrafish with spinal cord injury (Mokalled et al., 2016). The DUSP2 protein dephosphorylates the mitogen-activated protein kinase (MAPK) family, which regulates various cellular processes. Notably, several studies have demonstrated distinct roles of MAPK family members in axon regeneration. Previous reports showed that axon regeneration inCaenorhabditis elegans
requires the MAPK pathway (Li et al., 2012; Hisamoto et al., 2016; Cervellini et al., 2018). Dlk-1 MAPK kinase improves axon regeneration by activating the MAPK signaling cascade (Hammarlund et al., 2009; Yan et al., 2009). Other factors also influence regeneration by regulating the activation of the MAPK pathway inCaenorhabditis elegans
(Li et al., 2012; Byrne et al., 2014). Deletion of svh-1, which encodes a growth factor, damages axon regeneration ability because svh-1 plays an important role in activating the JNK cascade (Li et al., 2012). Because DUSP2 is a regulator of the MAPK signaling cascade, we suspected that DUSP2 may play a role in zebrafish axonal regeneration.The zebrafish spinal cord model is one of the ideal models for studying axonal regeneration. The spinal cord has a variety of distinct neurons, which innervate behavior of the organism (Tsata and Wehner, 2021; Varadarajan et al., 2022). Various models are used to study the regeneration of spinal cord injury, and the commonly used models are the contusion model and spinal cord crush (Shen et al., 2022; Varadarajan et al., 2022). However, these models cause a large lesion site and introduce the interference of other extrinsic factors. To minimize the interference of extrinsic factors and specifically study the function of DUSP2 on axonal regeneration, in this study, we used two-photon to perform axotomy, which causes limited lesion areas. Zebrafish larvae have transparent bodies and are easily targeted by two-photon axotomy; some studies have used two-photon axotomy to construct an axonal injury model in zebrafish (O’Brien et al., 2009; Hu et al., 2018; Sahu et al., 2018).
Mauthner cell (M-cell) is a motor neuron in the hindbrain of zebrafish, and the axon of M-cell projects to the tail of zebrafish along with the spinal cord (Burgess and Granato, 2007; Kohashi and Oda, 2008). We previously showed that the axons of M-cells can be completely regenerated over time after twophoton axotomy (Hu et al., 2018).
In this study, we constructed a homozygous mutation of dusp2 via CRISPR/Cas9 and examined nervous system development and motor function by micro-imaging and behavioral approaches. We used two-photon axotomy to injure the axons of M-cells and observed axonal regeneration after DUSP2 knockout, knockdown or overexpression. Our results show that DUSP2 negatively regulates the axonal regeneration of M-cells.
Methods
Animals
Adult zebrafish (Danio rerio
) and embryos were maintained at standard conditions (28.5°C, 14/10-hour light/dark cycle and sterilized water). We mated male and female adult zebrafish (3:2) overnight and collected embryos at dawn the next morning. Embryos were staged by days post fertilization (dpf). Larvae were housed in embryo medium containing 0.2 mM N-phenylthiourea (MilliporeSigma, Burlington, MA, USA), which prevents pigment formation (Milos et al., 1983). Wild-type (WT)/AB (China Zebrafish Resource Center, Wuhan, China, Cat# CZ1) and transgenic Tg (Tol 056) lines (RIKEN, Saitama, Japan), in which M-cells were labeled by green fluorescent protein, were used in this study. All experimental protocols were approved by the University of Science and Technology of China (USTC) Animal Resources Center and University Animal Care and Use Committee and the Committee on the Ethics of Animal Experiments of the USTC (approval No. USTCACUC1103013) in 2017.CRISPR/Cas9-mediated knockout and identification of
dusp2
mutants
Exon 1 of thedusp2
gene was selected as the Cas9-targeting region. We used mMessage mMachine T7 Ultra Kit (Thermo Fisher Scientific, Waltham, MA, USA) to synthesize Cas9 mRNA from suitable plasmids (43861; Addgene, Watertown, MA, USA) and the Megascript T7 kit (Thermo Fisher Scientific) to synthesize sgRNA from suitable plasmids (61051; Addgene). Cas9 mRNA anddusp2
guide RNA were mixed and co-microinjected into one-cell zebrafish embryos to obtain F0 larvae. The genomic DNA ofdusp2
gRNA-injected embryos and control embryos was extracted after 24 hours. Thedusp2
target fragment sequences were amplified by polymerase chain reaction andBgl
II (Takara, Dalian, China) was used to digest amplified fragments at 37°C for 0.5 hour. The mutant sequence cannot be digested by restriction endonucleaseBgl
II, while the wild-type sequence can be digested byBgl
II.F0 larvae were raised to adulthood and then crossed with AB zebrafish. Offspring (F1) were maintained to adulthood and crossed with their littermates for acquisition ofdusp2
zebrafish (F2). F2 adult zebrafish were screened byBgl
II digestion and Sanger sequencing (Sangon, Shanghai, China). DNA and protein sequence analysis were conducted by SnapGene3.1.4 (GSL Biotech LLC, San Diego, CA, USA). Tg(Tol 056);dusp2
zebrafish were constructed using the same process.Behavioral test
Free swimming of zebrafish larvae was examined to determine whether larval motor function changed after DUSP2 deletion. One-hour swimming distances were recorded and analyzed by Viewpoint equipment and software (Viewpoint, Lyon, France). The double -blind method was used to mitigate bias.
Single-cell electroporation
Thedusp2
coding sequence (CDS) was cloned by primers (Table 1
) and then inserted into upstream activating sequence (UAS) plasmids to construct the UAS-dusp2
plasmids. Following previously described methods (Chen et al., 2017), CMV-GAL4-VP16 (a plasmid that drives the expression of UAS plasmids), UAS-dusp2
plasmids and UAS-mCherry plasmids were cotransfected into unilateral M-cells at 4 dpf through single-cell electroporation for DUSP2 overexpression at the single-cell level. Healthy and morphologically normal larvae were selected and subjected to two-photon axotomy and imaging.
Table 1 |Primers used in this study
Two-photon axotomy
We used two-photon to injure the axon of M-cells. Zebrafish larvae (6 dpf) with M-cells expressing mCherry/green fluorescent protein were anesthetized in 100 mL embryo medium supplemented with 0.4% MS222 (Sigma) and then fixed in 1% low-melting agarose. We used a Zeiss 2-photon microscope (LSM710; Carl Zeiss, Oberkochen, Germany) to ablate the M-cell axons over the cloacal pores (O’Brien et al., 2009; Chen et al., 2017; Hu et al., 2018). A laser of 800 nm at an intensity of 20‒30% was used to ablate the M-cell axons.
In vivo
imaging of M-cells soma and axon regeneration
The M-cell soma and axon of anesthetized larvae were scanned with a confocal system (FV1000; Olympus, Tokyo, Japan) water immersion lens (340, 0.85 numerical aperture objective) for measurement of M-cell soma size at 4, 6 and 8 dpf and regeneration at 8 dpf. Adobe Photoshop CS4 (Adobe, San Jose, CA, USA) was used to evaluate images. To calculate the regenerated length, the point above the cloacal pores was selected as the start point. The length from the start point to the axonal terminal was defined as the total length. All fluorescent live images showed a lateral view of the spinal cord with the dorsal side above and the anterior to the left. We used ImageJ 1.50 (National Institutes of Health, Bethesda, MD, USA) (Schneider et al., 2012) to quantify the regeneration length. The double-blind method was used to mitigate bias.
Quantitative reverse transcription-polymerase chain reaction
Total RNA was extracted from AB anddusp2
larvae at 5 dpf using TRIzol reagent (Takara). The mRNA levels ofdusp2
,atf3
andklf6
were evaluated by quantitative reverse transcription-polymerase chain reaction (qRT-PCR) using specific primers (Table 1
) and the SYBR green kit (Invitrogen, Carlsbad, CA, USA). The reactions were performed in triplicate with three individual biological samples and three replicates following standard protocols (95°C for 5 minutes, followed by 45 cycles of 95°C for 10 seconds and 60°C for 30 seconds, and then 95°C for 15 seconds, 60°C for 60 seconds and 95°C for 15 seconds). Relative mRNA levels were obtained by normalizing toβ-actin
mRNA level using the 2method (Livak and Schmittgen, 2001). The doubleblind method was used to mitigate bias.Western blot assay
AB anddusp2
larvae at 5 dpf were lysed with radio-immunoprecipitation assay buffer (Sangon, Shanghai, China). Total proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane. The membranes were probed with the following primary antibodies: anti-p-JNK1/2/3 (rabbit; 1:1000; Abmart, Shanghai, China; Cat# TA3320S), anti-JNK1/2/3 (rabbit; 1:1000; Abmart; Cat# T55490S) and anti-β-actin (rabbit; 1:1000; HuaBio, Huangzhou, China; Cat# ET1701-80) for 4 hours at 26°C. The membranes were then incubated with secondary anti-rabbit (goat; 1:5000; Proteintech, Chicago, IL, USA; Cat# SA00001-2) at 26°C for 2 hours. ImageJ software was used to calculate the integrated optical density values. The double-blind method was used to mitigate bias.Statistical analysis
No statistical methods were used to predetermine sample sizes; we used a similar sample size to our previous work (Hu et al., 2018; Chen et al., 2019). The data were tested by normal distribution detection to determine whether they conformed to normal distribution. Data were analyzed with an unpaired, two-tailed Student’st
-test (normal distribution) or Mann-WhitneyU
test (non-normal distribution) using GraphPad Prism version 5.00 (GraphPad Software, San Diego, CA, USA, www.graphpad.com). Data are presented as the mean ± standard error of mean (SEM). All data were obtained from at least three repeated experiments. Statistical significance was set atP
< 0.05.Results
Construction of
dusp2
homozygous mutant zebrafish
We identified a target site indusp2
exon 1 and synthesized small guide RNA (sgRNA)in vitro
using this sequence (Figure 1A
). Cas9 mRNA and sgRNA were microinjected into the animal pole of zebrafish eggs at the first cell stage. F0 heterozygous mutant larvae were cultured to adulthood and crossed with wild-type AB to obtain the F1 generation. Homozygousdusp2
mutants were obtained after mating between F1 zebrafish littermates (Figure 1B
). We also constructed Tg(Tol 056);dusp2
zebrafish, whose M-cells express green fluorescence protein, following the same process (Figure 1B
).Sequencing results in F2 generation zebrafish revealed an 8 base pair deletion in exon 1 ofdusp2
that causes a frameshift mutation (Figure 1C
). We mated F2 homozygous mutants with AB and F2-self and confirmed the mutation by PCR and enzyme digestion. The results showed that DNA from thedusp2
heterozygous mutants, the offspring ofdusp2
and AB, showed both wildtype (resistant) and mutant (cut) fragments, while DNA from thedusp2
homozygous mutants, the offspring ofdusp2
-selves, showed cut fragments (Figure 1D
). Amino acid sequence analysis showed that the frameshift mutation caused a truncated DUSP2 protein lacking the rhodanese physiology domain and dual specific phosphatases cystine deletion, which renders DUSP2 nonfunctional as a phosphatase (Figure 1E
). These results confirm the successful construction ofdusp2
homozygous mutant zebrafish strains and show that the mutation is stably inherited.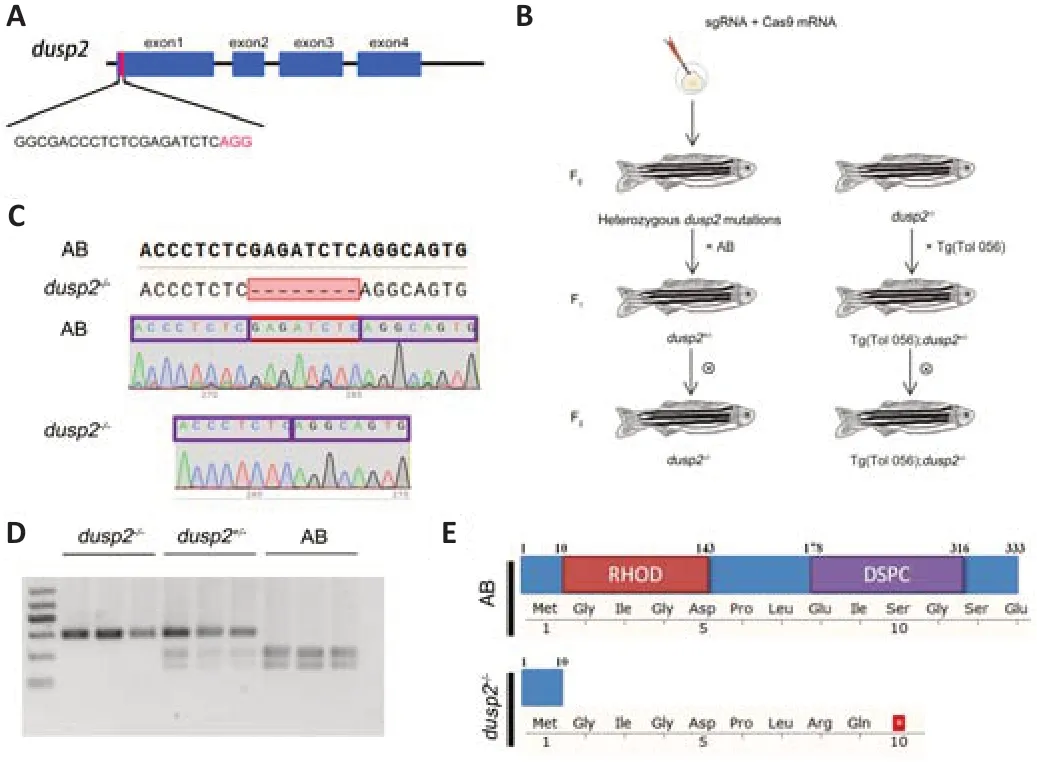
Figure 1 |Generation of DUSP2 knockout zebrafish using CRISPR/Cas9. (A) Schematic showing the Cas9-sgRNA-targeted site located in exon 1 of the dusp2 gene. (B) Generation of dusp2‒/‒ and Tg(Tol-056);dusp2‒/‒ zebrafish. (C) Sequencing results showed a frameshift mutation in dusp in the dusp2‒/‒ zebrafish caused by an 8 bp deletion. (D) Agarose gel results of DNA samples from AB, dusp2+/‒ and dusp2‒/‒ zebrafish digested with Bgl II. (E) Schematic of the protein sequence showing that DUSP2 mutated zebrafish expressed truncated DUSP2 composed of 10 amino acids, whereas wild-type DUSP2 is composed of 333 amino acids. DSPC: Dual specific phosphatases cystine; RHOD: rhodanese physiology domain; sgRNA: small guide RNA.
DUSP2 deletion does not affect M-cell development or motor function
We observed the M-cell soma larvae at 4, 6 and 8 dpf and counted the soma areas. In the absence of DUSP2, M-cell somas developed properly; there were no morphologic abnormalities in AB anddusp2
larvae (4 dpf:P
= 0.3346, 6 dpf:P
= 0.5106, 8 dpf:P
= 0.4142;Figure 2A
andB
). We then examined the total length of axons, defined as the axon length from the point above cloacal pores to the end in the tail. The total length showed no significant difference between AB anddusp2
larvae (4 dpf:P
= 0.0915, 5 dpf:P
= 0.6683; 6 dpf:P
= 0.5403;Figure 2C
andD
).To ascertain whether DUSP2 deletion affects larval nervous system function, we used free swimming, a behavioral test, to analyze the larval swimming distance over 1 hour. The results indicated that AB anddusp2
larvae had nearly the same total swimming distance during the examination time (P
= 0.9323;Figure 2E
andF
). Together, the imaging and free-swimming results showed that DUSP2 deficiency does not influence M-cell morphology and motor function in larvae. Our imaging results were consistent with a previous report (Maurer and Sagerström, 2018).Axon regeneration capacity is improved after dusp2 knockout
To examine the function of DUSP2 in axonal regeneration, we transected one of the M-cell axons at 6 dpf using two-photon axotomy. A round wound at the lesion site was found after two-photon axotomy (Additional Figure 1
).In vivo
imaging was then performed to investigate axonal regeneration of AB larvae anddusp2
larvae at 8 dpf. The imaging results indicated that regenerated axons indusp2
larvae were significantly longer than that of AB larvae (P
= 0.0025;Figure 3A
andB
). We then evaluated whether there was a gene dose effect between DUSP2 and axonal regeneration length. We found no significant difference in axonal regeneration length indusp2
larvae and AB larvae (P
= 0.4967;Figure 3C
andD
). These results indicate that DUSP2 is a negative regulator of axon regeneration and has no gene dose effect.DUSP2 overexpression retards axon regeneration
To acquire more information about DUSP2 function in axon regeneration, we constructed a UAS-dusp2 plasmids that overexpresses DUSP2 when cotransfected with CMV/Huc-GAL4-VP16 (Figure 4A
). We co-microinjected Huc-GAL4-VP16/UAS-dusp2
plasmids and Huc-GAL4-VP16/UAS-mCherry plasmids into one-cell stage eggs. Imaging results showed that mCherry was expressed in neuronal somata and axons (Figure 4B
). qRT-PCR results confirmed the overexpression ofdusp2
(P
< 0.0001;Figure 4C
).We then used single-cell electroporation to overexpress DUSP2 in M-cellsin vivo
. CMV-GAL4-VP16/UAS-dusp2
/UAS-mCherry plasmids and CMV-GAL4-VP16/UAS-mCherry plasmids were electroporated into 4 dpf larvae and labeled electroporated M-cells with mCherry fluorescent protein. Axotomy and imaging were performed at 6 and 8 dpf, respectively (Figure 4D
). We found that in comparison with axon regeneration in the control, axonal regeneration was retarded after DUSP2 overexpression (P
= 0.0181;Figure 4E
andF
). Taken together, these results revealed that DUSP2 overexpression leads to reduced axonal regeneration.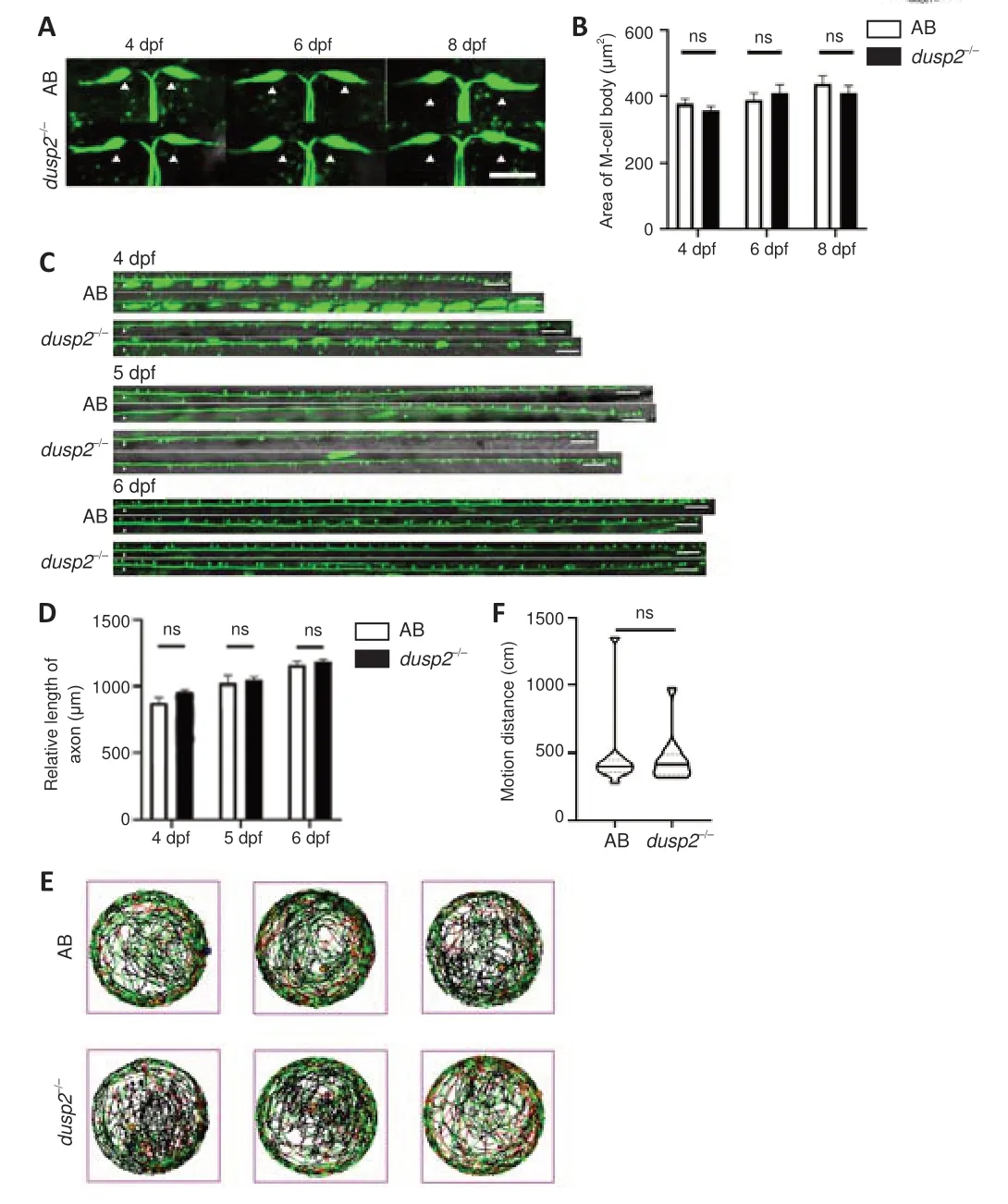
Figure 2 |DUSP2 knockout does not affect the development of M-cells or motor function. (A, B) AB (n = 18) and dusp2 homozygous mutants (n = 18) showed no significant difference in soma size. White triangle indicates the M-cell soma. (C, D) The total axon length in AB and dusp2 homozygous mutants was not different at 4 (n = 6), 5 (n = 6) and 6 dpf (n = 8). White triangle indicates the defined site. Scale bars: 50 µm. (E) The line portrays the swimming path of zebrafish larvae evaluated over 1 hour. Experiments were performed in a 24-well dish, with one larva in each well. Data for experimental (n = 12) and control (n = 12) groups comes from three groups of different AB parents and three groups of different dusp2‒/‒ parents. (F) Total movement distance of 1 hour in the freeswimming test. Soma size and defined total lengths of the axons were analyzed with unpaired, two-tailed Student’s t-test, and total movement distance was analyzed with Mann-Whitney U test. Error bars represent SEM. dpf: Days post fertilization; dusp2: dual specificity phosphatase 2; M-cells: Mauthner cells; ns: no significance.

Figure 3 |DUSP2 knockout enhances the regeneration capacity of M-cell axons.(A, B) Representative images of axon regeneration results in AB and dusp2‒/‒ (A) and statistical results (B) show that DUSP2 homozygous deletion increases axon regeneration length. (C, D) The axon regeneration length did not differ in AB and dusp2+/‒. White asterisk indicates the injury site. Scale bars: 50 µm. The axon regeneration lengths were analyzed with unpaired, two-tailed Student’s t-test. **P < 0.01; error bars represent SEM. DUSP2: Dual specificity phosphatase 2; M-cells: Mauthner cells; ns: no significance.
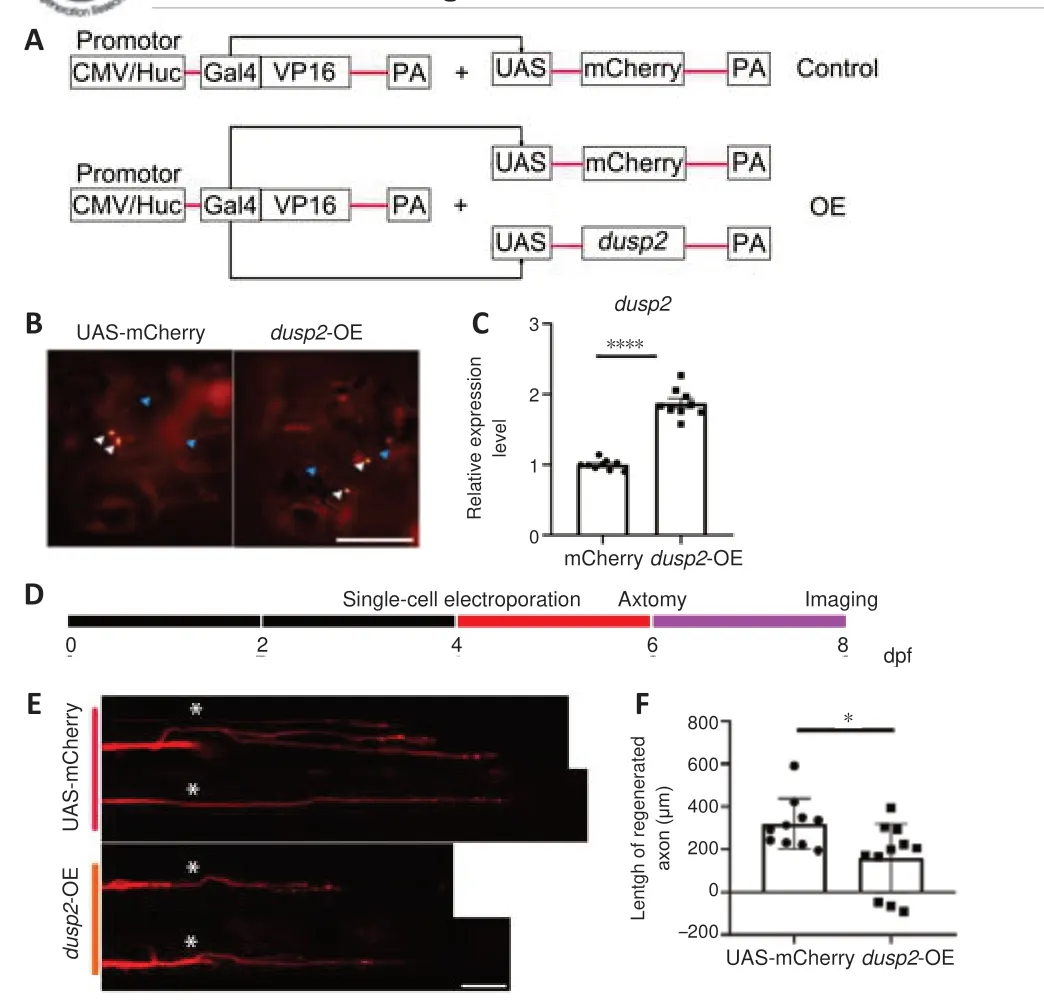
Figure 4 |DUSP2 overexpression weakens the capacity of axon regeneration. (A) Schematic diagram of the control and DUSP2 overexpression vectors. CMV/Huc-GAL4-VP16 plasmids drive expression of UAS-dusp2 plasmids and UAS-mCherry plasmids in all zebrafish cells or neurons. (B) Neurons of control larvae (injection of UAS-mCherry plasmids) and OE larvae (injection of UAS-dusp2 plasmids and UAS-mCherry plasmids) express mCherry fluorescent protein. White arrowheads indicate somata, and blue arrowheads indicate axons. Scale bar: 50 µm. (C) qRT-PCR analysis of dusp2. The total RNA of each group was extracted from 20 larvae at 4 dpf. Data were obtained from three independent experiments. (D) Schematic diagram of the experimental overview, with indicated time points of electroporation, axotomy and imaging. (E, F) Axon of OE larvae (injection of UAS-dusp2 plasmids and UAS-mCherry plasmids) shows retarded regeneration compared with control (injection of UAS-mCherry plasmids). White asterisk indicates injury site. Scale bar: 50 µm. The axon regeneration lengths and qRTPCR data were analyzed with unpaired, two-tailed Student’s t-test. *P < 0.05, ****P < 0.0001; error bars represent SEM. CMV: Cytomegalovirus; dpf: days post fertilization; DUSP2: dual specificity phosphatase 2; OE: overexpression; PA: poly adenylate; qRT-PCR: quantitative reverse transcription-polymerase chain reaction; ns: no significance; UAS: upstream activating sequence.
DUSP2 deletion increases JNK phosphorylation and activates downstream pathways to promote regeneration
A previous study reported that in DUSP2 deficient immune cells, JNK phosphorylation levels were increased (Jeffrey et al., 2006). To determine the effect of DUSP2 knockout on JNK phosphorylation in zebrafish, we used western blotting to detect JNK phosphorylation levels. While JNK protein levels were not changed after DUSP2 deletion (P
= 0.8055;Figure 5A
andB
), the level of p-JNK was increased after DUSP2 knockout (P
= 0.0378;Figure 5A
andC
). There was no difference in JNK mRNA level between AB and DUSP2 knockout (Figure 5D
). These results indicated that DUSP2 leads to decreased JNK phosphorylation in zebrafish.JNK and its downstream targets ATF3 and KLF6 have been reported to be correlated with axonal regeneration (Tedeschi and Bradke, 2013; Watkins et al., 2013; Schellino et al., 2019). ATF3 is upregulated after nervous system injury and drives axonal regeneration (Campbell et al., 2005; Renthal et al., 2020; Cheng et al., 2021). We found thatatf3
mRNA levels were significantly increased after DUSP2 deletion (P
= 0.0008;Figure 5E
). In contrast,klf6
, which was reported to promote axon regeneration (Kramer et al., 2021), was not affected (P
= 0.9595;Figure 5F
). These results suggest that DUSP2 deletion may promote M-cell axon regeneration by the p-JNK-ATF3 pathway.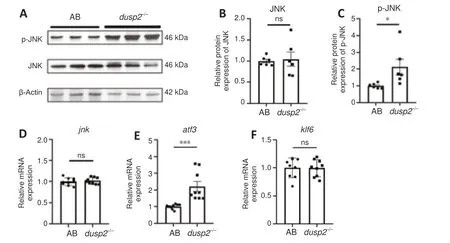
Figure 5 |DUSP2 knockout increases phosphorylation of JNK and activates downstream pathways. (A) Representative results of western blotting; data were obtained from three independent experiments, and each experiment had two replicates. The total protein of each group was extracted from 50 larvae at 5 dpf. (B, C) Quantification of JNK normalized to β-actin (B) and p-JNK normalized to JNK (C) from data shown in A. (D‒F) qRT-PCR analysis of jnk, atf3 and klf6 mRNA relative levels. The total RNA of each group was extracted from 50 larvae at 5 dpf. Data were obtained from three independent experiments. Western blotting and qRT-PCR data were normalized to results in the AB group, and data were analyzed with unpaired, two-tailed Student’s t-test. *P < 0.05, ***P < 0.001; error bars represent SEM. atf3: Activating transcription factor 3; DUSP2: dual specificity phosphatase 2; jnk: c-Jun N-terminal kinase; klf6: Krüppel-like factor 6; ns: no significance; qRT-PCR: quantitative reverse transcription-polymerase chain reaction.
Discussion
In this study, we found that DUSP2 knockout promoted axonal injury regeneration. We used single-cell electroporation to overexpress DUSP2 in zebrafishin vivo
and observed inhibited axon regeneration in the DUSP2 overexpression group. The DUSP2 phosphatase regulates JNK phosphorylation (Jeffrey et al., 2006), and the JNK pathway is involved in regulating the expression of various genes (Tedeschi and Bradke, 2013; Nguyen et al., 2015; Schellino et al., 2019). Our western blotting results demonstrated that DUSP2 deletion resulted in increased phosphorylation level of JNK. Bothatf3
andklf6
are downstream of JNK, and many studies have reported the role of ATF3 and KLF6 in axon regeneration (Campbell et al., 2005; Watkins et al., 2013; Renthal et al., 2020; Cheng et al., 2021; Kramer et al., 2021). Our qRT-PCR results showed that after DUSP2 knockout, the expression level ofatf3
but notklf6
increased significantly, which suggested that p-JNK-ATF3 may be a pathway affected by DUSP2. Our results revealed the function of DUSP2 in axon regeneration in zebrafish. Furthermore, our findings suggest that the p-JNK-ATF3 pathway may be the underlying mechanism of DUSP2 affecting axonal regeneration.DUSP2, also called PAC-1, was first cloned from human T cells and shown to exhibit phosphatase activity (Rohan et al., 1993). Farooq et al. (2003) showed the dynamic interaction between the C-terminal phosphatase domain of DUSP2 and the N-terminal domain of ERK2 of the MAPK family. Thisin vitro
result suggested that DUSP2 regulates the phosphorylation of ERK2 related sites (Farooq et al., 2003). Subsequent studies on its function in the immune system revealed that DUSP2 regulates JNK phosphorylationin vivo
(Jeffrey et al., 2006). JNK functions in axonal regeneration through phosphorylating different proteins such as neurofilament and STAT3 in the cytoplasm (Brownlees et al., 2000; Levy and Lee, 2002; Waetzig et al., 2006). Signal transducer and activator of transcription 3 (STAT3) is a crucial factor in the p-JNK-p-STAT3-ATF3/KLF6 signaling pathway and regulates axonal regeneration (Tedeschi and Bradke, 2013; Watkins et al., 2013; Schellino et al., 2019). Because of the lack of a specific antibody against zebrafish p-STAT3, we were not able to determine the involvement of activated STAT3 and future studies are required to assess whether STAT3 is upregulated when JNK phosphorylation level is increased by DUSP2 knockout.Phosphorylation of MAPK family members is vital for the nervous system. Few studies have examined the MAPK family members that influence zebrafish axon regeneration. The ubiquitin ligase phr controls the direction of axon regeneration through cypip2- and JNK-dependent pathways (Bremer et al., 2019). While the role of DUSP2 in the immune system has been investigated, few studies have focused its function in the nervous system. Our studies provide novel insights into JNK and DUSP2 function in the zebrafish central nervous system. Zebrafish larvae defective in DUSP2 did not show any changes in morphology and behavior. Previous studies also reported similar results (Maurer and Sagerström, 2018). Although DUSP2 may have little effect on the development of the central nervous system, it participates in various physiological processes in the central nervous system. Morente et al. reported DUSP2 function in granule neurons exposed to UV radiation and cisplatin. Their results revealed that P2Y13 receptors regulated DUSP2 expression, which prevented abnormal p38 activation, to protect granule neurons from UV radiation and cisplatin (Morente et al., 2014). In another study, researchers found decreased expression of DUSP2 in patients with major depressive disorder, and decreased DUSP2 led to MAPK overexpression (Martín-Hernández et al., 2018). These results revealed a protective effect of DUSP2 on neurons in abnormal situations related to MAPK overexpression, such as physical or chemical injuries and mental disorders. In our studies, we focused on the axonal regeneration of larvae and the JNK pathway‒induced, regeneration-increased atf3 expression. After DUSP2 deletion, enhanced phosphorylation of JNK increased ATF3 levels. Thus, we speculated ATF3 is involved in the mechanism of DUSP2 in axonal regeneration.
JNK regulates the expression of various downstream genes and performs different functions. ATF3 and KLF6, JNK targets, are upregulated after axon injury and promote axon regeneration (Campbell et al., 2005; Watkins et al., 2013; Renthal et al., 2020; Cheng et al., 2021; Kramer et al., 2021). Our results showed significantly increased expression of ATF3, but not KLF6, after DUSP2 knockout. The KLF family is an intrinsic factor closely related to neurite growth and axon regeneration (Moore et al., 2009). Different JNK subtypes regulate the expressions of different KLF family members; for example, KLF9 is regulated by JNK3 (Apara et al., 2017). We speculated KLF6 is not regulated by the JNK subtypes that DUSP2 dephosphorylate. In this study, we used antibody against p-JNK1/2/3, and more studies are needed to clarify the exact contribution of JNK1, JNK2 and JNK3.In conclusion, our results provide preliminary insights into the role of DUSP2 in axon regeneration and a potential mechanism. However, our research has several limitations. Our western blotting and qRT-PCR demonstrate that DUSP2 deletion increases the phosphorylation of JNK and expression of ATF3, both of which have been linked to axonal regeneration. Additional studies are needed to strengthen their involvement in the role of DUSP2, for example through knockdown of JNK and/or ATF3. More research is needed to determine the detailed mechanism of DUSP2. Notably, DUSP2 also regulates gene expression through epigenetic modifications (Dan et al., 2020). Therefore, further study is needed to clarify the upstream and downstream pathways of DUSP2 and determine these underlying mechanisms.
Author contributions:
Study design: GJS, DLR, BH; experiment implementation: GJS, XLW, MLW; data analysis: GJS, XLW; manuscript draft and revision: GJS, BH. All authors have read and approved the final version of the manuscript.
Conflicts of interest:
The authors declare no conflicts of interest.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:
Barbara Hausott, Innsbruck Medical University, Austria.
Additional files:
Representative diagram of axon successfully injured by two-photon.
Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Inflammation and retinal degenerative diseases
- Synaptic alterations as a common phase in neurological and neurodevelopmental diseases: JNK is a key mediator in synaptic changes
- Brain-derived neurotrophic factor in main neurodegenerative diseases
- The best of both worlds: mastering nerve regeneration combining biological and nanotechnological tools
- Exosomal miR-23b from bone marrow mesenchymal stem cells alleviates oxidative stress and pyroptosis after intracerebral hemorrhage
- Chlorogenic acid alleviates hypoxic-ischemic brain injury in neonatal mice