
有机-水混合溶剂中氯离子对C—H键的电氧化腈化性能
2022-08-10 09:50褚有群葛展榜焦玉峰张建平郭冠璇朱英红
化工学报 2022年7期
褚有群,葛展榜,焦玉峰,张建平,郭冠璇,朱英红
(浙江工业大学化学工程学院,浙江 杭州 310014)
引 言
有机电合成已成为一种重要的有机合成手段[1-2],凭借其绿色、高效的优点,已应用于阳极酰化[3]、卤化[4-5]、脱卤[6]、芳香醇的氧化[7]和乙烯醚聚合[8]等多种类型的有机合成反应中。间接电氧化是有机电合成中的一种重要方法,利用氧化电位更低的媒质,可避免反应底物直接在电极表面得失电子,并实现有机物在电解液中的均相催化转化[9]。氯离子作为一种无机媒质,因来源广泛、价格低廉引起了科研工作者的广泛关注。James 等[10]首次报道了以木糖为反应底物,通过氯离子电化学原位生成的HClO,一锅法脱氧合成δ-戊内酯。为避免乙烯在阳极上过度氧化,Leow 等[11]利用氯离子作为媒质,通过间接电氧化乙烯高选择性地合成环氧乙烷,此法在300 mA·cm-2的电流强度下电解100 h,仍能保持70%的电流效率。
在有机电化学合成中,大部分有机反应底物不溶于水溶液,因此需添加适量的有机溶剂形成有机-水混合溶剂。由于氯碱工业的重要性以及析氯过程受阳极材料、材料表面状态等因素影响的复杂性,许多研究者已经对氯化钠水溶液中氯离子的析氯过程动力学及反应机理做了大量的研究工作[12-14]。而在电化学反应体系中,溶剂对电化学性能影响很大,因此很有必要对氯离子在有机-水混合溶剂中的电化学性能进行系统研究。
以芳烃为原料,利用合适的催化剂降低反应的活化能将C—H键活化后,选择性地构建C—C、C—N等高附加值化合物可大幅缩短反应的合成路线,是一种高效、高原子经济性的合成手段[15-16]。如在芳香腈的合成路线中,甲基芳烃的气相氨氧化法是最主要的方法之一[17-18],该法通过对甲基上的C—H 进行活化,以氨气作为氮源,有效地避免了金属腈化试剂的使用,但是高温(>350℃)的反应条件对反应设备提出了更高的要求。电化学C—H 键活化法借助电势激活惰性底物,使得反应在温和条件下进行,为多种反应提供了新的合成思路[19]。Yoshida等[20]以吡啶作为氮源,实现了富电子芳烃中苯环上C—H键的电化学活化,此法和化学法中先硝化再还原的工艺流程相比,合成路线简单且无须添加催化剂。Onomura等[21]报道了一种电化学直接腈化法,以三甲基硅氰为氮源,借助阳极氧化激活环胺中α- C—H键可高选择性地合成α-氰基环胺。Pasha 等[22]发现以醛为底物,盐酸羟胺为氮源,ZnO 为催化剂,无须添加额外溶剂,可在微波条件下获得相应的腈。Movassagh 等[23]以乙二酰氯为脱水剂,直接将醛转化为相应的腈类化合物。
受上述醛化学转化为相应腈类化合物和本课题组在C—H 键电化学氧化为相应醛类化合物[24-25]的启发,本工作在有机-水体系中氯离子的电化学性能研究基础上,将氯离子的电化学转化性能应用于C—H 键的间接电氧化活化中,再进一步进行腈化反应合成相应腈类化合物,提出了一种在一室型无隔膜电解槽中,以氯离子为媒质,硫酸羟胺为氮源,通过电化学活化C—H 键原位生成醛,并进一步转化为相应腈类化合物的电化学合成方法。
1 实验材料和方法
1.1 材料
NaCl(西陇科学,纯度99.5%),对甲氧基甲苯(p-MeOBT,阿拉丁,纯度99%),四丁基高氯酸铵(TBAP,泰坦,纯度99%),乙腈、丙酮、甲醇、N,N-二甲基甲酰胺(DMF)、乙醚均为分析纯试剂,蒸馏水为实验室自制。
1.2 仪器
电解槽(50 ml 一室型夹套电解槽),CHI 660D 电化学工作站(上海辰华),多通道电化学工作站(Bio-Logic Science Instruments,法国),气相色谱仪(GC,Agilent 7890A,美国),DSQ Ⅱ单四极杆气质联用仪(GC-MS,Thermo FisherScientific,美国)。
1.3 电化学测试
循环伏安测试和线性扫描测试均在CHI 660D电化学工作站中进行,采用三电极体系,工作电极为Pt、GC、C 圆盘电极(直径3 mm),参比电极为Ag/AgCl电极,辅助电极为Pt电极(2 cm×2 cm),电解液为含有0.05 mol·L-1的TBAP 的有机溶剂-水混合溶液(除特别注明外,测试均在25℃下进行,p-MeOBT的浓度均为0.05 mol·L-1,NaCl的浓度均为0.05 mol·L-1)。
计时电量测试在多通道电化学工作站中进行,工作电极为Pt 圆盘电极(直径3 mm),参比电极为Ag/AgCl 电极,辅助电极为Pt 电极(2 cm×2 cm),电解液为含有0.10 mol·L-1的四丁基高氯酸铵的乙腈-水混合溶液,NaCl的浓度为0.01 mol·L-1。
恒电流电解在50 ml 一室型夹套电解槽中进行,阳 极 为Pt 电 极(2 cm×2 cm),阴 极 为Pb 电 极(2 cm×2 cm),电流密度为12.5 mA·cm-2。电解产物通过乙醚萃取分离后采用气质联用仪进行定性分析,采用气相色谱仪进行定量分析,分析方法为面积归一法。
2 实验结果与讨论
2.1 Cl-电化学氧化性能研究
2.1.1 不同电极上Cl-的电氧化行为 图1 为在乙腈-水混合溶液(体积比7∶3)中,Cl-分别在Pt、GC 和C 电极上的循环伏安图。由图可知,工作电极材料对Cl-的电化学氧化行为有明显的影响。当Pt 为工作电极时(曲线a),在1.42 V 和0.88 V (vs.Ag/AgCl)处,出现了一对氧化还原峰,表明在Pt 电极表面生成了一种能被还原的氧化态物质。以GC 作为工作电极时(曲线b),氯离子的氧化峰电位明显正移,和GC电极上空白溶液的CV 曲线(曲线d)比较,Cl-的氧化峰和溶液中的析氧峰出现部分重叠现象,不利于Cl-优先在电极上发生电氧化反应。而C 电极上析氧电位较低[26],氧化峰消失且起峰电位提前(曲线c),不利于Cl-的电氧化反应进行。

图1 Cl-在不同工作电极上的循环伏安图Fig.1 The cyclic voltammograms of Cl-on different working electrodes(v=50 mV·s-1)
2.1.2 不同有机溶剂和水混合溶剂中Cl-的电氧化行为 图2是在不同有机溶剂和水(体积比7∶3)的混合溶剂中,Pt电极上Cl-的循环伏安图。比较图中曲线可知,在有机溶剂和水混合溶剂的空白CV 曲线中,除了析氧峰外,没有出现其他氧化还原峰。在没有加入有机溶剂的NaCl水溶液中,析氧反应明显增强,但未出现Cl-的氧化还原峰。在质子型有机溶剂甲醇中也不利于Cl-的电氧化反应进行。而分别在非质子型有机溶剂丙酮、DMF 和乙腈的混合水溶液中,在1.4~1.5 V 电位区间出现Cl-的氧化峰,其中在乙腈-水混合溶剂中,Cl-的氧化峰电流最大。在该有机-水混合体系中,阳极上析氯和析氧反应是一对竞争反应,能溶于水的有机溶剂的加入可有效地包裹住水分子,促进Cl-在电极表面上优先发生电氧化反应。
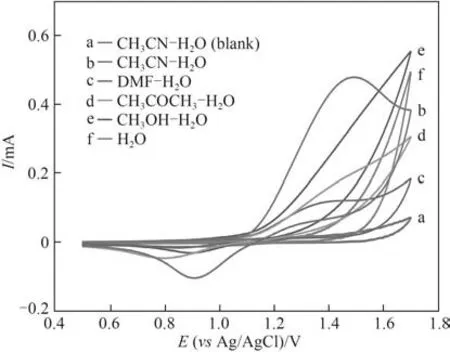
图2 Cl-在不同有机-水混合溶剂中的循环伏安图Fig.2 The cyclic voltammograms of Cl-in different organicaqueous solvents(v=50 mV·s-1,Pt electrode)
2.1.3 不同含水量时Cl-的电氧化行为 合适的非质子型有机溶剂和水的混合溶剂有利于Cl-优先在电极表面上发生电氧化反应,不同的有机溶剂与水的配比对Cl-的电氧化还原过程有很大的影响。不同含水量的乙腈-水混合溶剂中Cl-在Pt电极上的循环伏安图见图3。由图可知,随着含水量的增加,氧化峰电位均有所正移。但在含水量低于30%时,Cl-氧化峰电流明显上升,这说明适量的水能够促进反应进行。在高含水量(45%、50%),氧化峰电流以及还原峰电流明显下降,这可能是由于含水量过高,有机溶剂不足以包裹大部分水分子,使得水分子出现在电极表面的概率增大,增强了析氧反应和析氯反应的竞争。对比含水量分别为15%和30%时的氧化还原峰电流,30%含水量时Cl-的电氧化还原可逆性更好,因此后续实验在乙腈-水(体积比7∶3)混合溶剂中进行。

图3 Cl-在不同含水量乙腈-水混合溶剂中的循环伏安图Fig.3 The cyclic voltammograms of Cl-in different water contents of CH3CN-H2O solution(v=50 mV·s-1)
2.1.4 不同扫描速率下Cl-的电氧化行为 图4 是不同扫描速率下Cl-在Pt 电极上的循环伏安图。由图可知,在较低扫描速率(25 mV·s-1)时,循环伏安曲线中氧化峰较明显,还原峰电流很小。而随着扫描速率的增加,Cl-的氧化还原峰均明显增加。计算不同扫描速率下Cl-的氧化还原峰电流比|Iox/Ire|并列于表1中,发现随着扫描速率的增加,还原峰电流的增幅比氧化峰电流的增幅大,这是电化学-化学(electrochemical-chemical, EC)反应的典型特征,即在较低扫描速率时,电氧化反应后生成的中间体更易发生随后化学反应,同时随后化学反应的速率常数较小。随着扫描速率的增加,吸附在电极表面的吸附态中间体优先发生电化学转化,还原峰电流明显增大。这也说明在该有机-水体系中,Pt电极上Cl-的电氧化过程符合EC反应机理,这和Fiori等[27]提出的水溶液中Cl-的EC 反应机理类似,即Cl-首先在电极表面失去一个电子生成吸附态Clad,随后发生化学反应生成Cl2。同时在本有机-水溶液体系中,有利于Cl2在有水存在的环境中发生歧化反应,生成具有氧化性能的ClO-。

图4 不同扫描速率下Cl-的循环伏安图Fig.4 The cyclic voltammograms of Cl-at different scanning rates(v=50 mV·s-1)

表1 不同扫描速率下Cl-的氧化还原峰电流比|Iox/Ire|Table 1 |Iox/Ire|of Cl-at different scanning rates
2.2 Cl-电氧化动力学研究
2.2.1 表观活化能(Ea) 对于发生在金属/溶液界面上的电极反应,在不同温度下很难保证电极表面具有完全相同的表面状态和电极电位,因此可通过在恒定过电位下,不同温度时的电流密度求电极反应的表观活化能[28]:

图5 是在不同温度时Cl-的线性扫描图。由图可知,随着温度的提升,氧化峰电流不断增大,表明升高温度有利于Cl-的电氧化反应的进行。根据式(1),选取1.20、1.25、1.30、1.40 V (vsAg/AgCl)作为恒定过电位,对lgJ和1/T作图,得图6。由lgJ和1/T的线性拟合得到的直线斜率,可计算不同电位下Cl-的表观活化能Ea(列于表2 中)。在相同过电位差0.1 V 下,1.20~1.30 V(vsAg/AgCl)区间Cl-1的表观活化能下降3.887 kJ·mol-1,高于1.30~1.40 V 间表观活化能下降值(2.815 kJ·mol-1)。在电化学反应中,通常受扩散控制的反应活化能较低(8~21 kJ·mol-1),受电化学反应控制的反应活化能较高(>29 kJ·mol-1)[29],因此,在较低电位时,在该有机-水体系中Cl-的电氧化过程主要受电化学反应控制,而在较高电位下,则受电化学反应和扩散混合控制。
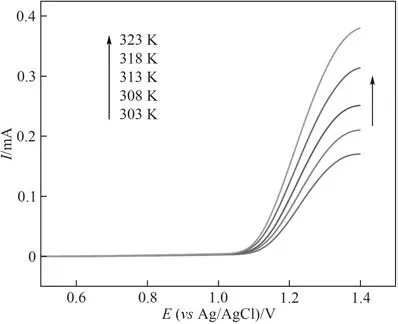
图5 Cl-在乙腈-水混合溶剂中不同温度下的线性扫描图Fig.5 The linear sweep of Cl-under different temperatures(v=10 mV·s-1)
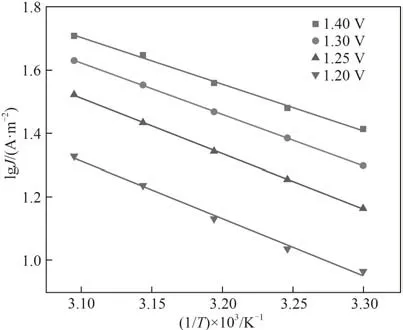
图6 lgJ与1/T的线性拟合图Fig.6 The relationship between lgJ and 1/T

表2 不同电位下Cl-电氧化反应的表观活化能Table 2 The apparent activation energy of Cl-at different potentials
2.2.2 扩散系数(D) 图7 是Cl-在不同含水量下的乙腈-水混合溶剂中的计时电量曲线。结合Cottrell方程[30]:
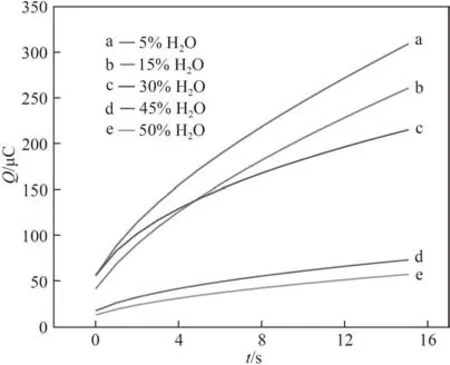
图7 Cl-在乙腈-水混合溶剂中不同含水量下的计时电量曲线Fig.7 Thechronocoulometry of Cl-in different water contents of the CH3CN-H2O solution
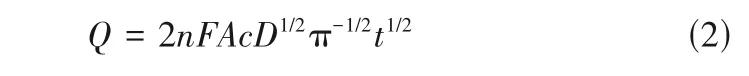
将Q与t1/2进行线性拟合,可得图8,通过拟合直线的斜率计算出不同含水量时的扩散系数D,结果见图9。由图可知,含水量对Cl-的扩散系数影响较大,且随着含水量增大其扩散系数明显下降。

图8 Q与t1/2的线性拟合图Fig.8 The relationship between Q and t1/2

图9 不同含水量的乙腈-水混合溶剂中Cl-的扩散系数Fig.9 The diffusion coefficient of Cl-in different water contents of the CH3CN-H2O solution
2.3 p-MeOBT电氧化腈化反应性能研究
2.3.1 Cl-对p-MeOBT 电氧化行为的影响 图10 是反应底物p-MeOBT 在乙腈-水(体积比7∶3)混合溶剂中的循环伏安图。由图可知,在不添加NaCl 时,p-MeOBT 起峰电位约为1.4 V,其氧化峰电位为2.1 V (vsAg/AgCl)。加入NaCl (曲线c)后,起峰电位有所负移,氧化峰电位明显负移[约1.9 V (vsAg/AgCl)],同时氧化峰电流明显增大,且还原峰消失,这说明Cl-对p-MeOBT 的电氧化过程具有明显的催化作用。

图10 p-MeOBT在乙腈-水混合溶液中的循环伏安图Fig.10 The cyclic voltammograms of p-MeOBT in CH3CN-H2O mixed solution(v=50 mV·s-1)
2.3.2 恒电流电解 表3 为在不同NaCl 浓度下,60℃,恒电流电解12 h,p-MeOBT 电氧化腈化反应的结果。当电解液中不添加NaCl (Entry 1)时,p-MeOBT 在电极表面直接电氧化,目标产物对甲氧基苯甲腈(p-MeOBCN)的收率只有28%。虽然反应底物的转化率达91%,但反应主要停留在中间产物对甲氧基苯甲醇(p-MeOBOH)、对甲氧基苯甲醛(p-MeOBA)和对甲氧基苯甲醛肟(p-MeOBO)阶段。当加入少量NaCl 后(Entry 2),p-MeOBCN 的收率有所提高,表明Cl-优先在电极表面发生电化学氧化反应,并进一步在水体系中生成具有氧化活性的ClO-,促进了对C—H 键的电氧化转化。但同时由于Cl-的加入,出现了另一含氯副产物3-氯-4-甲氧基苯甲腈(p-MeOBCl)。随着NaCl 浓度的继续增大(Entry 3,4),目标产物p-MeOBCN 的收率明显增加,当NaCl 浓 度 为0.15 mol·L-1时,目 标 产 物p-MeOBCN 的收率可提高到80%。而继续增大NaCl浓度达0.20 mol·L-1时(Entry 5),p-MeOBCN 的收率反而下降(71%),这可能是由于在高浓度的NaCl 下,体系中Cl-电氧化后的中间体浓度增加,使得含氯副产物也增加。

表3 不同NaCl浓度下p-MeOBT的电解结果Table 3 Electrolysis results of p-MeOBT in different concentration of NaCl
在Cl-电催化作用下,p-MeOBT 电氧化腈化反应过程中各物种的变化情况见图11。随着电解时间的增长(超过10 h),含氯副产物p-MeOBCl 的含量也逐渐增大。

图11 p-MeOBT电氧化腈化反应过程各有机物含量变化Fig.11 The contents change of each organics during electrolysis
2.3.3p-MeOBT 电氧化腈化的可能反应机理 根据电解反应过程中对中间物种的检测及反应机理推测,在Cl-电催化作用下p-MeOBT 间接电氧化腈化反应的可能机理如图12所示。
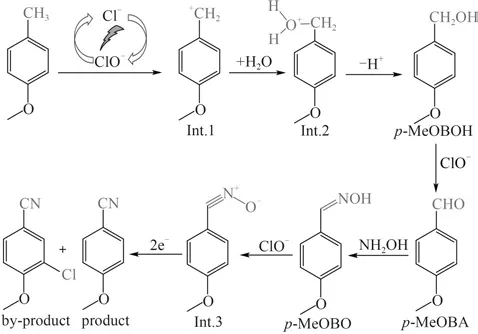
图12 Cl-作用下p-MeOBT间接电氧化腈化的可能反应机理Fig.12 Plausible reaction mechanism of the electro-oxidative cyanidation of p-MeOBT catalyzed by Cl-
3 结 论
(1)通过对有机-水混合体系的选择,有效保证了氯离子的电催化氧化性能。
(2)氯离子在乙腈-水混合溶剂中的电氧化反应属于EC反应机理,且第二步化学反应为控制步骤。
(3)氯离子在阳极上的电氧化过程在低电位时受电极反应控制,在高电位时受电极反应和扩散混合控制。因此在电合成反应时需要适量提高电流强度,确保氯离子在电极上的快速反应。
(4)在含0.05 mol·L-1TBAP 的乙腈-水混合溶剂中,60℃,电流密度为12.5 mA·cm-2条件下,恒电流电解12 h,p-MeOBCN的收率可达80%。
(5) 根据对中间产物的监控检测,提出了p-MeOBT间接电氧化腈化的可能反应机理。
符 号 说 明
A——工作电极面积,m2
B——常数
c——浓度,mol·L-1
D——扩散系数,cm2·s-1
Ea——表观活化能,kJ·mol-1
F——法拉第常数,C·mol-1
I——电流,A
Iox——氧化峰电流,A
Ire——还原峰电流,A
n——转移电子数
Q——电量,C
R——理想气体常数,J·mol-1·K-1
T——温度,K
猜你喜欢
四川水泥(2022年10期)2022-11-17
煤化工(2022年3期)2022-07-08
福建交通科技(2022年1期)2022-04-07
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
绿色科技(2020年18期)2020-11-05
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
首都食品与医药(2020年1期)2020-10-21
表面工程与再制造(2019年6期)2019-08-24
广东教育·高中(2018年12期)2018-02-13
山东工业技术(2016年10期)2016-09-06
