
植物天然产物氧化与微生物制造
2022-08-10 09:50:10张昕哲孙文涛吕波李春
化工学报 2022年7期
张昕哲,孙文涛,吕波,李春,
(1 北京理工大学化学与化工学院生物化工研究所,北京 100081; 2 清华大学化学工程系生物化工研究所/工业生物催化教育部重点实验室,北京 100084)
引 言
植物天然产物主要包括萜类、黄酮类、醌类、生物碱类等物质,具有丰富的生理药理活性[1-2]。在生成植物天然产物的过程中,氧化反应占重要地位,一般来说,氧化反应引入的羟基、环氧等官能团是天然产物后续进行糖基化、酰基化等许多其他修饰反应的前提,也是某些植物天然产物起关键药理活性的关键基团。如齐墩果烷型的三萜类物质β-香树脂醇,植物中的氧化酶可以在其28号碳位进行氧化生成抗菌药物齐墩果酸[3],也可以在11 号位和30 号位分别进行羰基化和羧基化,生成具有消炎和保护肝脏作用的甘草次酸[4],并可以通过后续的糖基化生成比蔗糖甜度高1000 倍的甜味剂单葡萄糖醛酸基甘草次酸和水溶性高于甘草次酸的甘草酸[5];黄酮类物质柚皮素碳骨架不同部位的氧化,可以生成圣草酚、花旗松素等具有不同药理活性的黄酮类物质[6]。此外,一些植物天然产物的氧化,还可以有效降低其细胞毒性。例如,来自喜树的具有抗癌活性但具有较高细胞毒性的生物碱类化合物喜树碱,在其10位进行羟化后得到10-羟基喜树碱,在提高抗癌活性的同时成功降低了喜树碱对细胞的毒性[7],成为目前广泛使用的一种抗癌药。
植物天然产物的氧化反应大多是通过生物体内的氧化酶催化进行的。氧化酶的种类丰富多样,包括黄素依赖的氧化酶、酪氨酸酶、漆酶、P450 酶等[8-10]。为实现氧化反应的进行,氧化酶大多需要辅基参与,如黄素、金属离子、血红素等,这些辅基参与电子、质子等物质的传递。此外,在氧化反应发生过程中需要质子与电子供体。参与氧化反应的电子来源较为多样,可来源于辅酶、辅基或底物本身[11]。参与催化氧化的辅因子成本往往较为高昂,而生物体中富含氧化反应所需的辅因子,因此目前合成生物学领域利用氧化酶催化氧化反应常以生物体作为依托进行[12]。
合成生物学为微生物利用氧化酶催化植物天然产物氧化提供了有效方法[13]。将植物天然产物合成途径包括氧化酶转入微生物体内,可实现植物天然产物的合成。由于植物生长较慢需要长时间占用耕地,并且植物天然产物往往结构复杂,所以相比于传统获取植物天然产物的植物提取法和化学全合成法,微生物酶催化合成植物天然产物具有高效性、高立体选择性和高区域选择性的优势[14]。
由于氧化反应对生产植物天然产物的重要性,本文首先将介绍具有代表性的萜类、生物碱、黄酮等植物天然产物的氧化。按照氧化酶中所含辅基的差异,分类介绍催化植物天然产物发生氧化作用的氧化酶,并介绍不同辅基催化氧化反应进行的反应机理。此外,本文还将介绍目前在合成生物学领域氧化酶在萜类、生物碱、黄酮等植物天然产物合成中的应用和促进氧化酶高效行使氧化功能的方法。最后,对未来氧化酶在微生物合成植物天然产物领域的前景进行展望。
1 植物天然产物形成中的氧化反应
按照化学结构,植物天然产物主要分为萜类、甾体、糖苷、黄酮、生物碱、醌类。本节着重对萜、黄酮和生物碱类化合物碳骨架的氧化作举例说明。
1.1 萜烯化合物骨架的氧化
萜烯物质是目前在生物体中合成途径较清晰且广受关注的一类重要天然化合物。天然的萜烯物质主要由甲羟戊酸途径(MVA)和2-甲基-D-赤藓醇-4-磷酸途径(MEP)生成的五碳单元进一步反应生成[15-16]。
在萜类物质碳骨架的氧化反应中,引入的含氧官能团主要有羟基、羰基和羧基等。氧化是生成萜类物质的重要反应,如单萜物质柠檬烯经过一步氧化形成薄荷烯醇和香芹醇,进一步将其羟基氧化分别形成薄荷烯酮和香芹酮[17];在倍半萜物质青蒿酸的合成过程中,紫穗槐-4, 11-二烯的C-12 需经过第一步氧化反应生成青蒿醇,随后经过第二步氧化反应生成青蒿酮,并经过最后一步氧化反应生成青蒿酸[18];二萜物质松香二烯经过生成醇、醛、酸的三步连续氧化反应,最终生成松香酸[19]。
三萜物质的碳骨架更加复杂,按照碳骨架的区别可将三萜物质细分为齐墩果烷型、乌苏烷型、羽扇豆烷型等。值得注意的是,这些环状三萜都是以角鲨烯为起始物,经过角鲨烯环氧化酶的环氧化作用形成2,3-环氧角鲨烯,再被环化酶环化而形成,2,3-环氧角鲨烯中的2,3-环氧基团最终形成环状三萜C-3 的羟基。齐墩果烷型三萜的代表性物质β-香树脂醇C-28可依次进行羟基化、醛基化、羧基化,最终形成具有消炎作用的齐墩果酸[3];除此之外,β-香树脂醇的C-11 经历羟基化、羰基化后生成11-羰基-β-香树脂醇,随后在其C-30 发生羟基化、醛基化以及羧基化,最终生成具有保护肝脏作用的甘草次酸[5];乌苏烷型的三萜物质α-香树脂醇,在其C-28 先后发生羟基化、醛基化以及羧基化后,生成具有抗癌作用的熊果酸[20];羽扇豆烷型的三萜物质羽扇豆醇,经过C-28的三步连续氧化后生成具有显著抗癌功效的白桦脂酸[21]。萜类物质的氧化不仅提高了萜类物质的水溶性使之更容易在生物体中进行代谢和转运,同时也为后续的糖基化、酰基化修饰提供了条件,例如甘草次酸在C-3 羟基的基础上进行糖基化修饰生成单葡萄糖醛酸基甘草次酸、甘草酸[22]等。
具有代表性的萜类物质氧化过程如图1所示。
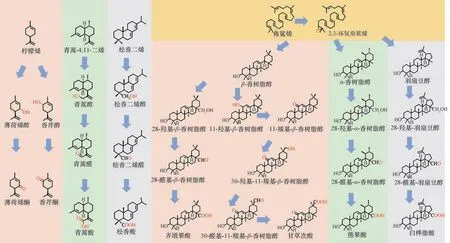
图1 具有代表性的萜类物质氧化过程Fig.1 Oxidation process of representative terpenes
1.2 黄酮类化合物骨架的氧化
黄酮化合物广泛存在于自然界中,莽草酸途径是其主要的生物合成途径[23]。黄酮类物质的基本母核是2-苯基色原酮。按照结构来划分,黄酮类天然产物主要包含黄酮、异黄酮和查尔酮等。目前,生成黄酮和异黄酮类化合物的氧化反应报道较多。
2-苯基色原酮母核氧化会产生丰富的黄酮类物质。例如,在2-苯基色原酮的5,7,4′位羟化得到芹菜素,继续在3′位发生氧化生成具有抑制癌细胞增殖作用的木犀草素[24];或在芹菜素的基础上在3 号碳位继续发生氧化生成具有抗癌、抗癫痫、抗炎作用的山奈酚,进一步在3′位发生氧化生成低毒性、抗氧化、抗肿瘤的槲皮素[25];在5,7-二羟基黄酮基础上,6 号碳位进行氧化生成能够治疗心脑血管疾病的黄芩素;以二氢黄酮代表性物质柚皮素为骨架,在3,5′进行氧化可生成有清除自由基作用的二氢槲皮素[26]。异黄酮类物质,具有抗氧化作用的大豆素,需要在异黄酮母核的7,4′发生羟基化;若继续在大豆素的基础上进行C-6 羟化以及转甲基化,生成能够改善人体糖、脂代谢的鸢尾素。根皮素需要在异查尔酮母核2′,4′,6′,4 位发生羟基化并还原中央三碳原子的不饱和双键[27]。
黄酮化合物碳骨架氧化引入的羟基也为后续的糖基化、甲基化等反应提供了条件。如在槲皮素的C-3羟基引入葡萄糖基和鼠李糖基可生成具有抗炎、抗氧化、抗过敏、抗病毒等功效的芦丁;在鸢尾素C-7 羟基引入葡萄糖基可获得水溶性更好、生物利用度更高的鸢尾苷;在根皮素C-6′羟基引入葡萄糖基后获得能有效治疗糖尿病的根皮苷[28]。
具有代表性的黄酮物质骨架的氧化如图2所示。
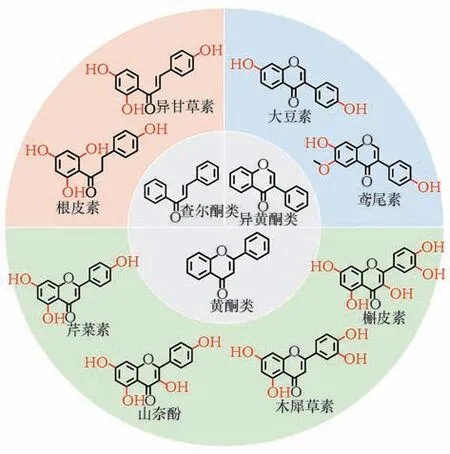
图2 具有代表性的黄酮物质骨架的氧化Fig.2 Oxidation process of representative flavonoids
1.3 生物碱类化合物骨架的氧化
生物碱是一类分布广泛的天然化合物,现指存在于生物体中的含氮环状化合物。此类化合物因普遍含有负氧化态的氮原子而往往呈碱性。由于结构的多样性,生物碱在生物体中具有更加复杂多样的合成途径[29]。
生物碱的氧化赋予其丰富的药效。例如来自颠茄的托品烷类莨菪碱,在其托品烷的C-6、C-7 发生环氧化反应会形成东莨菪碱;若只在莨菪碱C-7发生羟基化反应则形成山莨菪碱,东莨菪碱和山莨菪碱均可起到胃肠道解痉、抑制唾液分泌、镇静和扩瞳的作用;若在阿托品的α碳发生羟基化则生成樟柳碱,具有抗震颤、解痉、平喘、散瞳、抑制唾液分泌及解毒的功能。喹诺里西啶类生物碱苦参碱,在其N-1发生氧化形成氧化苦参碱,相比于苦参碱,氧化苦参碱具有更好的抗乙肝病毒的作用。来自蓝果树科植物喜树的喜树碱是一种被广泛用于治疗胃肠道癌的喹啉类生物碱,在起到抑癌活性的同时也会引起高度毒性的药物不良反应,在其10号碳位发生氧化引入羟基可以有效地减轻这种副作用,同时提升其抑癌功效[7]。苯菲啶类生物碱二氢血根碱和二氢白屈菜红碱,在其氮杂环氧化脱氢会分别生成血根碱和白屈菜红碱,均具有抗真菌增殖的功能。
生物碱碳骨架的氧化会为后续的生物碱修饰提供更多契机。如阿扑吗啡C-11 发生羟化后进而转移甲基生成木兰碱;C-9 羟化后向C-9 羟基转移甲基生成氧化海罂粟碱。
具有代表性的生物碱骨架与其氧化产物如图3所示。

图3 具有代表性的生物碱骨架与其氧化产物Fig.3 Oxidation process of representative alkaloids
2 氧化酶的种类及其催化机理
植物天然产物骨架的氧化,离不开生物体中结构和功能多样的氧化酶。氧化酶包括黄素依赖的单加氧酶、单胺氧化酶、过氧化物酶、P450 酶等种类,因为氧化酶在行使氧化作用的同时涉及氧气、电子以及质子的传递,因此往往需要辅酶和辅基等辅因子的参与。同种辅基催化氧化反应机理相似,因此本节按照氧化酶中所含辅基的种类对其进行分类,并对氧化反应的催化机理进行阐释。
2.1 辅因子为黄素的氧化酶
辅因子为黄素的黄素依赖型单加氧酶(FMOs)可参与氧化结构复杂的天然产物[11],这些反应源于其催化中心黄素异咯嗪环体系多样的化学特性[30-31]。除此之外,蛋白质在调节黄素辅因子的催化潜力方面也起着重要作用,因为游离黄素本身并不能执行这些反应[32-33]。了解和预测黄素辅因子活性如何受到蛋白质环境的调节仍然是一个热门的研究话题[32]。
目前已经用实验和计算相结合的方法研究了FMOs 的黄素动力学、反应物种的形成和稳定以及FMOs 的羟基化机理(图4):首先,处于还原状态下的黄素被氧气激活,并将一个电子从还原的黄素转移到氧中,在黄素半醌和超氧阴离子之间形成笼状自由基对,随后超氧阴离子与黄素结合,并伴随着质子的传递,形成黄素C4a-(氢)氧加合物[34];根据中间体质子化状态不同,这种过氧基团与底物反应使氧-氧键断裂,同时异咯嗪远端的氧插入底物的C—H键,而另一个黄素近端的氧原子被还原为水,有研究表明异咯嗪C-8 位被亲电取代基取代会加速氧-氧键断裂[35],且目前已经利用羟基邻位具有高电负性的对羟基苯甲酸进行了实验和理论计算,证明底物中若有高电负性区域的碳会更容易进攻C4a-(氢)氧加合物(过氧黄素)的氧-氧键,从而实现底物中此碳位的羟基化[36-37];随后黄素形成氧化状态的黄素,需要NADPH 等辅因子提供的还原力以及质子将氧化态的黄素还原为还原状态下的黄素,催化下一轮氧化反应[38],
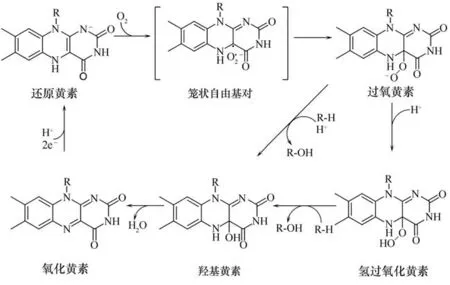
图4 黄素单加氧酶催化氧化机理Fig.4 Mechanism of flavin-dependent monooxygenase catalytic oxidation
黄素依赖型单加氧酶基于其结构特征、氨基酸序列、电子供体和氧化反应类型进行分类[9]。目前已报道的具有代表性的黄素单加氧酶共有八类[11],其依赖的辅因子、电子供体以及催化的反应各不相同。表1对黄素依赖的单加氧酶分类进行了概述。
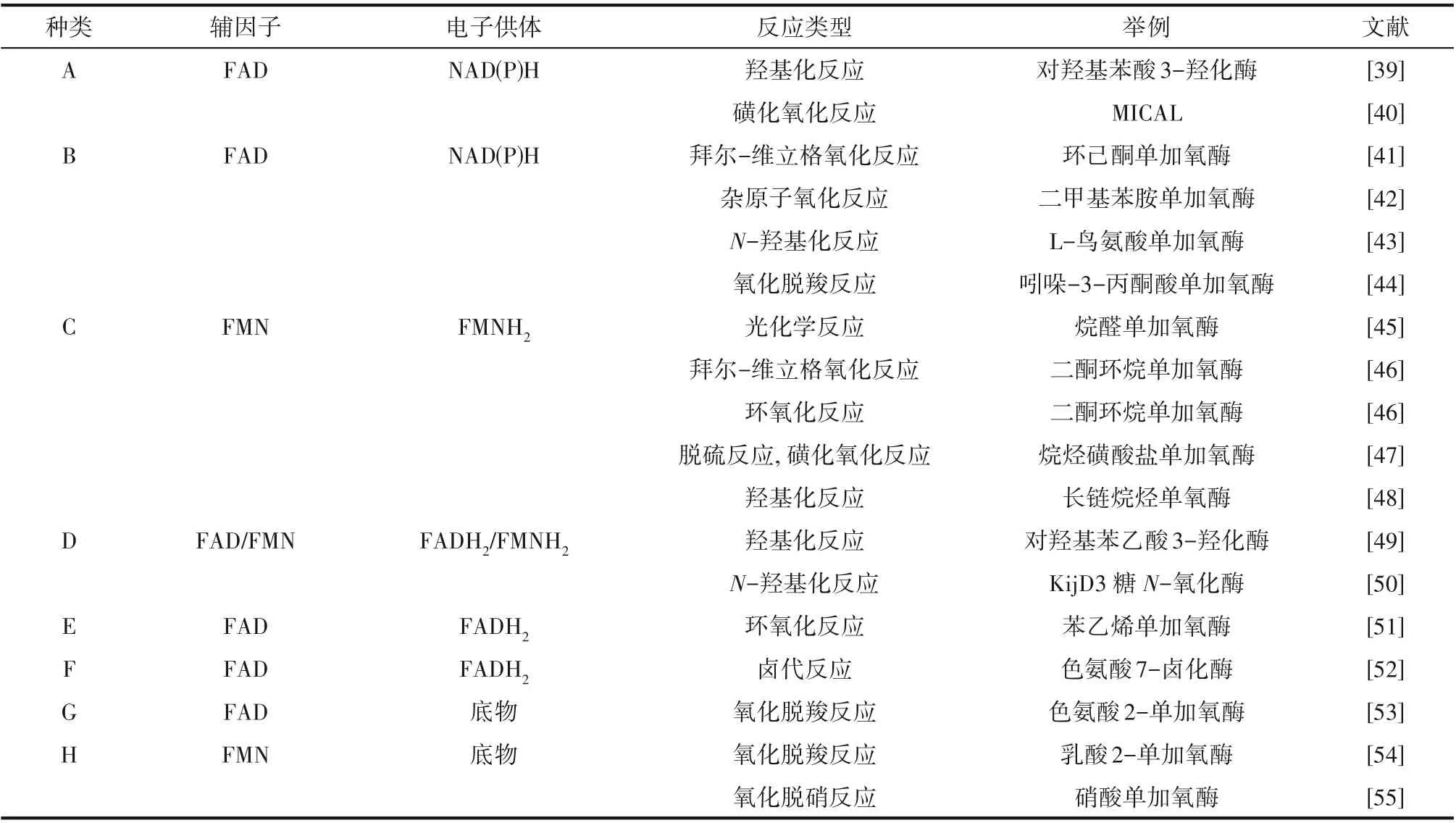
表1 黄素依赖的单加氧酶主要类别[11]Table 1 Main classes of flavin-dependent monooxygenases[11]
作为一种具有代表性的黄素依赖的氧化酶,依赖FAD 的单胺氧化酶(MAOs)因具有高选择性和活性,以及酶促合成所固有的环境优势,广受生物催化方面的关注和报道[56]。MAOs 以氧气分子为氧化剂,将胺氧化脱氢形成亚胺[57]。配合单胺氧化酶的氧化脱氢作用可以利用后续的化学反应将亚胺进行加成反应,形成伯胺的对映异构体,也可以利用亚胺进行继续脱氢形成叔胺[58],因此MAOs 是可以用于氧化多种不同胺并进行手性胺拆分的高性能生物催化剂。
2.2 辅因子为铜离子的氧化酶
一些氧化酶活性中心含金属离子,这些金属离子是酶的关键辅因子,对整个催化过程起控制作用,这类酶被称为金属酶。在氧化酶中,以铜离子为辅因子的氧化酶较为常见,包括铜蓝蛋白、酪氨酸酶、漆酶、抗坏血酸氧化酶等。
隶属于多铜氧化酶家族的铜蓝蛋白是一种以铜离子为辅基的氧化酶。铜蓝蛋白在发挥催化氧化过程中铜离子中心会接受来自底物提供的电子,随后进一步将电子传递给氧分子,使之还原为水或过氧化氢[59]。另外铜蓝蛋白对机体内的苯二胺还能发挥胺氧化酶作用[60]。
具有双核铜离子结构的多亚基氧化酶酪氨酸酶(Tyr),对酪氨酸等苯酚类化合物具有催化邻位氧化的功能。酪氨酸酶的双核铜离子Cu(A)Cu(B)和与两铜离子配位的六个组氨酸残基构成了酪氨酸酶的活性中心。在酪氨酸催化的整个氧化过程中,铜离子、氧分子、酪氨酸三者会形成多种反应中间体与过渡态(图5)。这些中间体过渡态主要被分为氧化态(Eoxy)、还原态(Emet)、脱氧态(Edeoxy)和失活态(Edeact)四种。

图5 来自巨型芽孢杆菌的酪氨酸酶及其活性位点组氨酸与铜离子的配位(PDB:3NM8)Fig.5 Tyrosinase from Bacillus megaterium and coordination of its active site histidine with copper ion(PDB:3NM8)
酪氨酸酶催化苯酚类化合物邻位氧化的整个循环过程如图6 所示:首先酚羟基氧负离子靠近活性中心并与一个铜离子配位,同时酚羟基邻位碳靠近与铜离子配位的一个氧;与此同时,Cu(A)配位的一个组氨酸咪唑氮接受由酚羟基解离出质子[10,61-63]从而使Cu(A)与组氨酸解离;Cu(A)和与其配位的过氧桥一起靠近苯酚的邻位碳原子,利于苯酚邻位碳原子进攻过氧桥;随后活性中心的[O—O]键断裂,靠近苯酚邻位的氧与高电负性的苯酚邻位结合,使氧与苯酚邻位成键从而形成复合物E;复合物E 再接受两个质子,与活性中心解离生成邻苯二酚;邻苯二酚可以为下一步氧化反应提供电子,自身被进一步氧化为邻苯二醌,与此同时处于还原态的酪氨酸酶(Met)被还原成脱氧态的酪氨酸酶(Deoxy);脱氧态的酪氨酸酶在接下来的反应中可以直接与氧分子结合,使氧化态的酪氨酸酶(Oxy)重新生成,催化下一轮的氧化反应的进行。目前酪氨酸酶已被用于生物合成多种化合物,其反应过程的动力学也被广泛研究[64]。
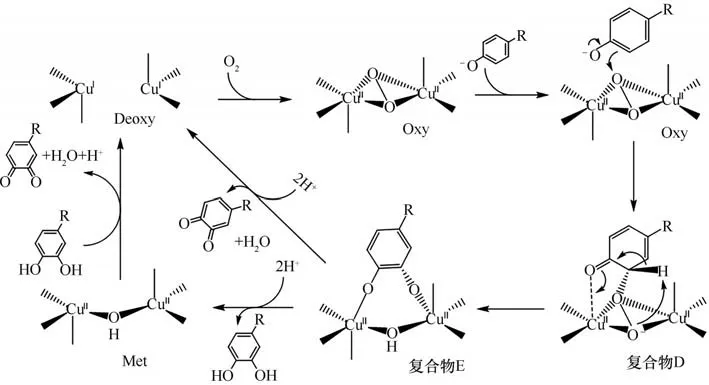
图6 酪氨酸酶催化酚类物质氧化机理[61]Fig.6 Mechanism of phenolic oxidation catalyzed by tyrosinase[61]
2.3 含血红素的氧化酶
过氧化物酶和P450 酶是两种典型的利用血红素作为辅基催化氧化反应进行的氧化酶。
过氧化物酶可以催化底物的氧化反应,同时将电子传递给过氧化氢,使之还原为水。过氧化物酶可以通过上述过程清除生物体内的过氧化氢以及胺类和酚类化合物,从而减轻它们对细胞造成的毒性。近年来,过氧化物酶在氧化催化中的应用得到了广泛的综述[65-67]。
细胞色素P450 酶也是一个含血红素的蛋白家族,血红素铁通过轴向保守的半胱氨酸与蛋白质相连[8,68]。P450 在天然产物的生物合成途径、外源物质的降解、类固醇激素的生物合成和药物代谢中发挥重要作用。P450 被认为是自然界中用途最广泛的生物催化剂,参与20 多种不同类型的化学氧化反应[69-71]。
P450 催化系统主要包含底物、P450 酶、起电子传递作用的P450 还原酶以及提供还原力的辅因子NAD(P)H 四个部分[8,72]。这四个部分使P450 酶催化氧化反应顺利进行。以P450 催化的羟基化反应为例(图7),P450中包含静息状态铁的活性中心首先接受底物,底物取代活性中心的水分子,但底物并不直接与铁结合。然后,高自旋状态的铁(FeⅢ)接受来自其氧化还原伴侣的电子被还原为亚铁(FeⅡ)。随后,氧气进入酶的活性位点并与FeⅡ结合形成[FeⅡO2]配合物。配合物[FeⅡO2]被来自氧化还原伴侣的第二电子还原形成配合物[FeⅢO22-],它利用溶剂中的质子生成过氧铁配合物[FeⅢ-OOH],被称为化合物0。伴随第二个质子进入活性位点,[FeⅢ-OOH]的O—O 键断裂,释放出一个水分子,生成高价卟啉自由基阳离子四价铁[FeⅣ=O](即化合物Ⅰ)。这种高活性的配合物易从底物中争夺一个氢原子,形成铁基羟基化合物[FeⅣ-OH](即化合物Ⅱ)。随后,伴随底物自由基与化合物Ⅱ的羟基反应生成羟基化产物(R-OH),铁也由化合物Ⅰ中的四价被还原为三价。最后,一分子水返回活性位点与FeⅢ配位并将底物置换,使FeⅢ恢复静息状态。当底物分子再次进入P450 活性口袋并将水分子置换,使FeⅢ变为高自旋状态时,相同的催化循环被再一次启动。值得注意的是,一些P450能够直接利用H2O2作为电子供体,直接使高自旋状态的FeⅢ形成化合物0,并利用过氧化物分流途径行使催化功能(图7,虚线箭头)。然而,除了P450 过氧化物酶(如CYP152 亚家族)[73]外,大多数P450的低效和低H2O2耐受性极大地限制了这种分流途径的应用。

图7 P450羟化机理Fig.7 Hydroxylation mechanism of P450
值得一提的是,P450 催化循环的维持依赖于氧化还原伴侣向血红素铁的持续电子传递,这是个复杂的电子传递系统。根据氧化还原伴侣的类型和P450与氧化还原伴侣的相互作用关系,P450氧化还原系统可以分为五个主要类别[74-76](图8)。大多数细菌和线粒体P450 中存在的第Ⅰ类P450 系统具有一个双组分的氧化还原伴侣系统,这种系统由一个含FAD 的铁氧还蛋白还原酶(FDR)和一个含铁硫的铁还蛋白(FDX)[77]组成。真核生物使用的Ⅱ类P450系统有一个单组分的氧化还原伴侣,这种氧化还原伴侣是一种膜结合蛋白,其含有的FAD和FMN将来自NADPH 的电子传递给血红素,被称为细胞色素P450还原酶(CPR)。Ⅲ类P450系统有一个真核细胞样的CPR,并通过一个柔性连接子融合在P450血红素结构域的C 末端。Ⅳ类P450 系统以FMN/Fe2S2还原酶结构域为电子供体,FMN/Fe2S2还原酶结构域与P450结构域通过连接子连接。除此之外,一些P450可以直接与它们的电子供体相互作用,并且不依赖于额外的氧化还原伙伴蛋白来完成催化反应,被划分为第Ⅴ类P450。Ⅲ~Ⅴ类P450 不依赖于氧化还原伴侣蛋白,通常被称为自给自足的P450。
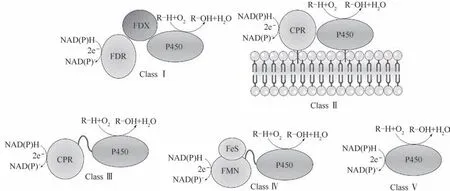
图8 五个主要类别的P450氧化还原系统[76]Fig.8 Five main categories of P450 REDOX systems[76]
3 微生物制造植物天然产物的氧化过程
氧化酶在天然产物合成过程中起着至关重要的作用。且酶催化反应的高效性、高立体选择性和区域选择性赋予了酶高效合成手性药物的能力[78],因此利用氧化酶催化合成天然产物有着植物提取、化学全合成无法替代的优势。
3.1 萜类化合物合成过程中的氧化酶
近些年,植物中的氧化酶,尤其是P450 酶催化的萜烯氧化反应广为报道。例如,CYP71AV9 可以将倍半萜类物质吉玛烯A 氧化为吉玛烯酸,吉玛烯酸进一步被CYP71BL5 氧化为6-羟基吉玛烯酸,作为洋蓟内酯生物合成途径的前体物质,为洋蓟内酯的生物合成奠定基础[79];在紫苏中挖掘的CYP76AJ1在酵母细胞中被证实具有氧化补身醇形成水蓼二醛的功能,为其生物合成提供借鉴[80];CYP701A26被鉴定为能够氧化二萜化合物ent-贝壳杉烯生成赤霉素前体物质ent-贝壳杉烯酸的P450 酶,是赤霉素生物合成过程中的重要一步[81];CYP716 家族的P450氧化酶绝大多数具有氧化各类三萜化合物C-28 的作用[82],例如CYP716A179 可以专一地氧化各种三萜化合物的C-28 位:将羽扇豆醇28 号碳位氧化为羧酸生成具有显著抗肿瘤活性的桦木酸[83],将β-香树脂醇28 号碳位氧化为羧酸生成对急性肝损伤有明显保护作用的齐墩果酸[84],将α-香树脂醇的28号碳位氧化为羧酸生成具有对革兰阳性菌、革兰阴性菌以及酵母菌有明显抑制作用的熊果酸[85],同样CYP716A44 和CYP716A46 也 具 有 相 同 的 功 能[86]。CYP716 家族的另一种P450 氧化酶CYP716A175 还能氧化日耳曼醇的28 号碳位形成羧酸,生成模绕酸[87]。CYP716 家族的CYP716A141 除了能氧化β-香树脂醇的C-28,也能氧化其C-16,在其对应位置生成羟基,合成马尼拉二醇[88]。除此之外,CYP716A1、CYP716A2 也具有相似的氧化功能[89]。来自番茄的CYP716E26 可以特异性氧化β-香树脂醇C-6 生成曼陀罗萜二醇[86]。CYP716C49 可以氧化齐墩果酸的C-2,生成山楂酸,氧化熊果酸C-2 生成科罗索酸,氧化桦木酸C-2 生成麦珠子酸[90]。CYP714 家族的CYP714E19 可以氧化齐墩果酸的C-23生成丝石竹酸;同时也可以氧化乌苏烷型的五环三萜C-23 生成相应的羧酸[91]。CYP87 家族的P450氧化酶CYP87D18可以专一性氧化葫芦二烯醇的11 号碳位,在对应位置生成羟基,随后继续催化羟基氧化为羰基[92]。来自甘草的CYP88D6,可以催化β-香树脂醇C-11 氧化生成甘草次酸的前体物质11-羰基-β-香树脂醇[93]。来自苜蓿的CYP72A68 和CYP72A67 分别连续地对齐墩果酸的C-2、C-23 进行氧化,分别生成常春藤素和贝萼皂苷元,随后CYP72A68 还可以继续对常春藤素C-23 进行连续氧化,生成棉根皂苷元和丝石竹酸[94]。来自苜蓿的CYP72A63 和来自甘草的CYP72A154,可以氧化β-香树脂醇和11-羰基-β-香树脂醇的C-30,分别生成11-脱氧甘草次酸和甘草次酸[4]。
3.2 生物碱合成过程中的氧化酶
生物碱的合成途径的复杂多样造成了生物碱碳骨架的多样化,与此同时生物碱的氧化修饰也随之变得更加丰富,参与生物碱氧化反应的氧化酶种类也更繁多,其氧化反应类型也更加多样。
Hagel 等[95]鉴定了黄素蛋白氧化酶DBOX 和TPOX,分别能催化二氢血根碱和四氢罂粟碱氧化脱氢,生成血根碱和罂粟碱;Ghislieri等[96]利用来自黑曲霉的单胺氧化酶突变体(MAO-N)催化的去消旋反应,催化(±)-毒芹碱生成(R)-毒芹碱,也可利用突变体催化(±)-胡秃子碱和(±)-细茜花碱分别去消旋化形成(R)-胡秃子碱和(R)-细茜花碱,同样他们也发现这种单胺氧化酶的突变体能够催化四氢-β-咔啉环系的去消旋化,得到去消旋化的四氢-β-咔啉环[97],是带有四氢-β-咔啉环生物碱微生物合成的关键一步;Xu等[98]发现FMN 依赖的氧化酶PhzG 能够催化吩嗪类化合物碳骨架的氧化脱氢;Meng等[99]鉴定了来自链霉菌S.sapporonensis的6 种氧化酶的功能,其中5 种是依赖于酮戊二酸/Fe2+的双加氧酶(BcmB、BcmC、BcmE、BcmF、BcmG),另一种是细胞色素P450 单加氧酶(BcmD),这六种氧化酶通过羟基化、氧化脱氢、环氧化,将环磷酰胺最终氧化为双环霉素;Ju等[100]在茄病镰刀菌Fusarium solani中鉴定了一种D-氨基酸氧化酶FsDAAO,该氧化还原酶可高度特异地将四氢异喹啉类化合物去消旋化,催化其氧化脱氢并进而反应生成(S)-四氢异喹啉类化合物;Couturier等[101]发现契珠哈氏菌Hahella chejuensis中的黄素依赖氧化酶PigB 和HapB,经鉴定这两种酶可催化2-甲基-3-戊基二氢吡咯生成2-甲基-3-戊基吡咯,作为灵菌红素的重要前体物质,这两种氧化酶的发现对灵菌红素的生物合成具有借鉴意义。
3.3 黄酮化合物合成过程中的氧化酶
目前,因氧化是植物类黄酮生物合成的重要步骤,黄酮类天然产物氧化酶的挖掘也成为了热门话题[102]。黄酮合成过程中的第一个羟基化反应由P450 酶C4H 催化,在C-4 位引入一个羟基从肉桂酸到A环,形成对香豆酸[103]。目前,多种黄酮氧化酶的功能已经被解析。催化黄酮骨架氧化的酶主要是P450 酶,如大豆苷元分别在氧化酶nfa12130 和nfa33880 的氧化作用下,可以分别在苷元A 环的C-6、C-8 位进行氧化生成相应的羟基[104];大豆苷元也可以在P450 酶CYP107H1 的氧化作用下,在其B 环 的C-3′ 形 成 羟 基[105];P450 酶SAV2377、SAV5841、SAV4539 对染料木素、白杨素、芹黄素中B环具有较好的氧化活性[106]。
3.4 其他天然产物合成过程中的氧化酶
近些年,醌、糖苷和甾体类天然产物合成途径的氧化酶也有报道。
Ehrlich 等[107]在黄曲霉素合成基因簇中发现了一个氧化酶HypC,催化降散盘衣酸蒽酮C-10 氧化为降散盘衣酸;Throckmorton 等[108]验证了烟曲霉中的氧化酶TpcL,催化大黄素蒽酮C-10 氧化为大黄素;Marzorati 等[109]利 用 来 自 毛 栓 菌Trametes pubescens的漆酶氧化葡萄糖及其衍生物生成葡萄糖醛酸,Baratto 等[110]利用同样的漆酶将硫代秋水仙碱苷、苦杏仁苷、积雪草皂苷和人参皂苷的葡萄糖苷氧化为葡萄糖醛酸;Ncanana 等[111]也利用来自Trametes pubescens的漆酶氧化桃柘酚的酚羟基形成自由基中间体,进一步反应生成二聚桃柘酚;甾体的合成往往也需要氧化酶参与催化氧化脱羧[112],也需要P450氧化酶对其骨架进行氧化,如Kiss等[113]报道来自巨大芽孢杆菌Bacillus megaterium的CYP106A1可催化强的松醇的C-11羟基和C-15,生成15β-羟基强的松,也可以催化地塞米松C-11 的羟基氧化,生成11-羰基地塞米松。
表2列举了部分天然产物氧化物催化实例。

表2 天然产物氧化酶催化举例Table 2 Examples of natural product oxidase catalysis
4 微生物细胞工厂中氧化过程的调控
在天然产物合成中,氧化反应的进行往往会受到一些因素的制约。例如:氧化还原反应伴随的辅因子消耗、氧化酶在行使催化作用时电子等物质的传递耦合性低和传递效率低等,使氧化酶经常成为级联反应中制约物质转化效率的限速酶,这也是目前微生物合成天然产物面临的瓶颈问题。如何解决这一瓶颈问题,是实现氧化酶高效催化天然产物氧化的关键。微生物制造天然产物的氧化过程调控方法主要可分为酶工程、代谢工程和底物工程等。
通过代谢工程以及酶工程提高氧化酶在生物体内的活性的方法是目前合成生物学中的研究热点(图9)。挖掘活性更高的同工酶、氧化还原伴侣适配、增强辅因子的供应、酶的定点突变、定向进化等是目前较为常用且有效的策略[114]。

图9 氧化酶氧化过程调控的主要方法Fig.9 Main methods for regulating the oxidation process of oxidase
作为典型的氧化酶,P450 酶的电子传递是它的瓶颈问题。研究表明,P450与CPR 的有效适配能够增强电子传递,进而增强其氧化效率。由于乌拉尔甘草根部积累大量甘草次酸,而叶片几乎没有甘草次酸积累,因此Zhu 等[115]通过分析乌拉尔甘草转录组中甘草根与叶片的转录组差异,挖掘到一个P450氧化还原伴侣GuCPR1,酵母细胞体内验证发现GuCPR1 可与CYP88D6 和CYP72A63 高度适配,使甘草次酸在酿酒酵母内的产量达到(18.9±2.0)mg/L,适配效果远远强于来自苜蓿和拟南芥的CPR;Kim 等[116]利用酿酒酵母异源表达原人参二醇的合成途径,研究P450 酶与CPR 的适配,在表达来自人参的P450氧化还原伴侣PgCPR5后人参二醇产量达到最高,使原人参二醇的产量提高了2.5 倍,证明PgCPR5与人参二醇合成途径中的P450有着更强的适配性。
融合蛋白的策略也给氧化酶的活性提升提供了参考。Mellor 等[117]利用铁氧还蛋白与模式植物高粱的CYP79A1 融合,催化蜀黍苷生物合成的第一步。与铁氧还蛋白的融合使CYP79A1 能够通过直接与光系统Ⅰ相互作用获得用于催化的电子。此外,融合的铁还蛋白部分捕获的电子被更有效地导向P450催化活性,使融合能够更好地与耦合到代谢途径的内源电子汇竞争。P450-铁氧还蛋白融合酶仅从融合的铁氧还蛋白获得还原力,在体内表现优于未融合的CYP79A1,表明电子从光系统Ⅰ到CYP79A1 的转移大大增强。Wang 等[118]利用融合蛋白的策略融合级联反应的两种酶,将1,8-桉叶油素合成酶(CS)和能在1,8-桉叶油素C-6 形成羟基的CYP176A1 两酶融合,使单萜1,8-桉叶素的羟基化程度提高了5.4倍。
除此之外,对P450血红素结构域和氧化还原伴侣CPR 的理性改造也是提升P450 氧化能力的一个有效手段。Sun 等[119]采用定点突变的策略,调整CYP72A63 内 部 的 疏 水 环 境,CYP72A63T338S突 变 体提高了CYP72A63 氧化11-羰基-β-香树脂醇C-30的能力,CYP72A63L509I使CYP72A63 专一氧化11-羰基-β-香树脂醇C-30 形成羟基,CYP72A63L398I使CYP72A63 专一氧化11-羰基-β-香树脂醇C-29 生成羟基。
5 总结与展望
氧化反应是天然产物合成过程中普遍存在的反应类型。植物天然产物骨架后修饰中的氧化反应是提高其在生物体内水溶性的重要保障,也是天然产物进行后续的糖基化、酰化等反应的前提。氧化反应使天然产物种类更丰富、药理活性更多样。近些年人们对天然产物的氧化反应进行研究实验,打通了众多天然产物的生物合成途径,也开发出许多药理活性明显优于天然产物的天然产物衍生物。
氧化酶是催化天然产物进行氧化反应的重要催化剂,它具有高区域选择性、高立体特异性和高催化活性的特点。目前,氧化酶在微生物细胞工厂中的应用仍受到许多制约,主要包括:(1)氧化还原反应的实质是电子的得失,微生物体主要利用辅因子向氧化酶传递电子,因此微生物表达外源氧化酶基因会消耗大量辅因子,若微生物细胞中辅因子供给不足,则会严重制约氧化酶催化的氧化反应进行;(2)微生物细胞内环境与植物细胞内环境存在差异,植物中的氧化酶于微生物细胞内异源表达的表达量、氧化酶在异源宿主内表达的稳定性问题,也是制约氧化酶高效、长时间行使其催化功能的因素;(3)许多氧化酶因为自身的杂泛性,催化后所得产物繁多,造成目标产物得率低,进而给目标产物的分离纯化带来一定的困难;(4)有些氧化酶在催化氧化过程中会产生活性氧等对微生物细胞有害的物质,对宿主微生物自身代谢带来强烈的干扰,严重干扰微生物本身的生存和繁殖,制约植物天然产物的微生物制造。利用代谢工程强化辅因子供应、增强宿主微生物的耐受性、利用酶工程的策略改造氧化酶,从而提高其催化特异性、催化活性和热稳定性是目前的研究热点,也是解决氧化酶在微生物体内高效发挥氧化作用的重要方法。
酶工程、代谢工程与合成生物学的快速发展,使微生物实现高效氧化反应进而生产天然产物成为可能。将酶工程与微生物代谢工程、发酵工程等手段有机结合,实现天然产物的高效合成,是未来天然药物合成的发展趋势。
猜你喜欢
昆明医科大学学报(2020年12期)2021-01-26 00:44:12
海峡姐妹(2019年8期)2019-09-03 01:01:02
天然产物研究与开发(2019年1期)2019-03-01 05:41:22
天然产物研究与开发(2018年1期)2018-02-02 07:21:19
中成药(2017年12期)2018-01-19 02:06:58
中成药(2017年12期)2018-01-19 02:06:32
中成药(2017年7期)2017-11-22 07:33:25
中成药(2017年8期)2017-11-22 03:18:58
中国粮油学报(2016年5期)2016-01-23 02:44:40
中国当代医药(2015年24期)2015-03-01 02:06:12
