
致病疫霉诱导的马铃薯酵母双杂交文库构建及无毒蛋白PiAVR3b寄主靶标筛选
2022-08-08 03:56栾宏瑛王荟洁王洪洋陈爱娥
植物保护 2022年4期
韦 吉, 栾宏瑛, 王荟洁, 刘 晶, 王洪洋*, 陈爱娥
(1. 云南省马铃薯生物学重点实验室, 云南省高校马铃薯生物学重点实验室, 云南师范大学, 昆明 650500; 2. 云南师范大学教务处, 昆明 650500)
马铃薯Solanumtuberosum起源于南美洲安第斯山山脉一带,是解决全球粮食短缺和消除贫困的重要作物。然而在生长过程中,马铃薯会受到各种各样病虫的危害,其中由卵菌中致病疫霉Phytophthorainfestans引起的晚疫病是马铃薯生产上最具毁灭性的病害。全球每年因马铃薯晚疫病造成的经济损失高达数十亿欧元,马铃薯晚疫病已对世界粮食安全构成了严重威胁[1]。
为了成功侵染寄主植物,病原菌会分泌大量效应蛋白到植物细胞内以干扰植物免疫系统。与大豆疫霉Phytophthorasojae、橡树疫霉P.ramorum基因组相比,致病疫霉不仅基因组大(约 240 Mb),而且还含有更多的分泌型效应子基因,RxLR胞质效应子(R:精氨酸;x: 任意氨基酸;L:亮氨酸)就是其中一类。通过对致病疫霉的基因组分析,发现有563个RxLR效应子基因。这些 RxLR 效应蛋白大多扮演着毒性因子的角色,致病疫霉通过吸器组织将其分泌到寄主细胞内干扰植物免疫反应,从而使致病疫霉能够快速侵染并在寄主植物中繁衍[2]。当寄主植物细胞内有抗病蛋白(resistance protein, R)时,R蛋白会特异性识别对应的RxLR类效应蛋白,从而激发ETI免疫反应(effector-triggered immunity)。然而,RxLR效应蛋白除激发植物 ETI免疫反应外,更多的功能是抑制病原相关分子模式激发的 PTI(PAMP-triggered immunity)或 ETI免疫反应信号传导,从而大大减弱植物的抗性反应[3-8]。
酵母双杂交(yeast two-hybrid)技术是一种直接于酵母细胞内检测蛋白质-蛋白质相互作用且灵敏度很高的分子生物学方法。其原理是利用真核生物转录因子(GAL4等)的DNA结合功能域(DNA binding domain, DNA-BD)和转录激活结构域(activation domain, DNA-AD),将待研究的病原菌效应蛋白和寄主靶标蛋白分别与BD和AD结构域连接并共转化进入酵母菌中,若效应蛋白和寄主靶标蛋白存在互作,则转录因子的BD和AD结构域相互靠近,即可转录激活下游报告基因的表达,以此来检测这2种蛋白互作的发生[9]。Du等[10]通过酵母双杂交证实致病疫霉无毒蛋白AVR1与囊泡运输相关蛋白Sec5互作进而促进致病疫霉侵染寄主。Boevink等[11]利用酵母双杂交发现效应蛋白PITG_04314与植物蛋白磷酸酶 PP1c 催化亚基互作,并促进 PP1c从核仁转运到核质,但是 PITG_04314 并没有影响 PP1c 的磷酸酶活性。研究者推测 PITG_04314 是通过与 PP1c 发生互作形成PITG_04314-PP1c复合体全酶形式负调控植物防御反应。Turnbull等[12]利用酵母双杂交发现致病疫霉无毒蛋白AVR2靶向3个BSL家族成员(BSL1,BSL2,BSL3),通过调控油菜素内酯信号传导促进致病疫霉侵染。同年,He等[13]通过酵母双杂交证实致病疫霉效应蛋白PiSFI3/Pi06087/PexRD16与U-box激酶 StUBK发生互作以抑制植物PTI免疫。可见,酵母双杂交在致病疫霉效应蛋白与寄主靶标蛋白研究方面有广泛应用。
PiAVR3b是一个典型的RxLR类效应蛋白。当寄主植物含有对应抗病蛋白R3b时,PiAVR3b可作为无毒蛋白被R3b识别并激发ETI免疫反应[14]。Zheng等[4]报道PiAVR3b可以抑制Flg22介导的早期PTI免疫反应。Wang等[15]发现 PiAVR3b能够抑制致病疫霉效应蛋白 PITG_22798 介导的细胞死亡反应。实验室前期证实,瞬时表达PiAVR3b显著促进致病疫霉侵染本氏烟。上述研究表明,PiAVR3b在调控植物免疫反应方面发挥着重要作用。但是,PiAVR3b如何参与调控植物免疫及寄主靶标蛋白尚不清楚。本研究构建了受致病疫霉诱导的马铃薯叶片cDNA文库,并以PiAVR3b为诱饵筛选获得4个寄主靶标蛋白,为进一步揭示PiAVR3b调控植物免疫分子机理提供依据。
1 材料与方法
1.1 材料
马铃薯栽培种‘合作88’种植于云南师范大学马铃薯科学研究院温室。致病疫霉菌株88069(1.3.4.7)、PIC99183(1.3.4.5.7.8.10.11)由华中农业大学马铃薯课题组馈赠。对‘合作88’,PIC99183是毒性菌株;88069是无毒菌株,将两菌株混合后接种‘合作88’用于构建马铃薯酵母双杂交cDNA文库。PIC99183用于PiAVR3b基因克隆。根据候选互作靶标基因编码序列(coding sequence,CDS),利用Primer 3在线软件(https:∥bioinfo.ut.ee/primer3-0.4.0/)进行引物设计。引物由生工生物工程(上海)股份有限公司合成。
酵母Y2HGold菌株、酵母Y187菌株、pGADT7、pGBKT7、pDONR222、pGADT7-DEST、大肠杆菌感受态细胞DH5α、DH10B、“pGADT7-T+pGBKT7-53”(1对强互作蛋白,后文简称53+T)、“pGADT7-T+pGBKT7-Lam”(1对不互作蛋白,后文简称Lam+T)购自上海欧易生物科技有限公司。具体载体信息、用途及构建方法见酵母双杂交使用手册(Clontech,PT4084-1)。高保真酶、反转录试剂盒、无缝克隆试剂盒购自南京诺唯赞生物科技股份有限公司。DNA纯化回收试剂盒、植物总RNA提取试剂盒、酵母质粒提取试剂盒购自北京天根生物技术有限公司。酵母缺陷培养基SD/-Leu、SD/-Trp、SD/-Leu/-Trp、SD/-Leu/-Trp/-His、SD/-Leu/-Trp/-His/-Ade、SD/-Trp/-His/-Ade、YPD Plus液体培养基、X-α-Gal、鲑鱼精DNA购自TaKaRa公司。CloneMiner Ⅱ cDNA 构建试剂盒、FastTrack MAG mRNA分离试剂盒、反转录试剂盒等购自Invitrogen公司。
1.2 方法
1.2.1致病疫霉诱导的马铃薯叶片cDNA文库构建
致病疫霉88069菌株和PIC99183菌株在黑麦固体培养基(每升含有黑麦60 g,蔗糖20 g,CaCO31.2 g,琼脂15 g)上生长15 d左右,往长满白色菌丝及孢子囊的培养皿里加入无菌水(3 mL/皿),用玻璃棒轻刮培养基表面菌丝,调整孢子囊浓度至104个/mL。将2个菌株孢子囊悬浮液等体积混合后接种‘合作88’离体叶片,取接种后24 h和48 h的叶片各20片,分别采用TRIzol 法提取两个时间点总RNA并进行等量混合,用DNaseⅠ消化酶去除残留DNA后检测总RNA浓度和纯度,利用1%琼脂糖凝胶电泳检测其完整性。参照FastTrack MAG mRNA分离试剂盒和CloneMinerⅡcDNA合成试剂盒说明书,分离mRNA反转录成cDNA第一链,并合成cDNA第二链。将双链cDNA与attB1和attB2重组接头连接(attB1-cDNA insert-attB2)后进行分离与浓缩纯化,利用 BP重组反应(25℃连接16~20 h)将纯化的cDNA和含有 attP1和attP2的载体pDONR222连接。将重组产物电转化大肠杆菌DH10B(1 500 V,200 Ω,25 μF),在转化体系中加入 4 mL SOC培养基225~250 r/min振荡培养1 h,涂布平板,次日用SOC培养基洗板3次,100 mL无菌离心管收集菌液,往离心管中加甘油至终浓度20%并保存于-80℃,即为初级文库菌液。从初级文库菌液中提取质粒,将质粒与pGADT7-DEST进行 LR 重组反应(25℃连接16~20 h),将重组产物纯化后电激转化大肠杆菌DH10B,在转化体系中加入4 mL SOC培养基225~250 r/min振荡培养1 h,取样涂布平板确定库容量;剩余培养物加入甘油至终浓度 20%存于-80℃,即为次级文库菌液。随后从次级文库菌液中提取质粒,将5 μg次级文库质粒转化进入Y187酵母菌株中,用于后续酵母双杂交。
1.2.2cDNA文库容量、重组率及插入片段长度的鉴定
取初级、次级文库细菌原液10 μL稀释100倍,分别取50 μL初级、次级文库稀释液涂布于含50 mg/L Kan和50 mg/L Amp的LB固体培养基平板上,第2天计数鉴定库容量。文库菌液库容量(cfu/mL)=平板上的克隆数/50 μL×100倍×1 000 μL。文库总库容量=文库菌液库容量×文库菌液总体积(mL)。从各级文库中随机挑取24个克隆进行菌落PCR鉴定。初级文库菌落PCR鉴定引物为M13(F: 5′-GTAAAACGACGGCCAG-3′; R:5′-CAGGAAACAGCTATGAC-3′);次级文库菌落PCR鉴定引物为pGADT7-DEST (F: 5′-TAATACGACTCACTATAGGGCGAGCGCCGCCATG-3′; R: 5′-GTG-AACTTGCGGGGTTTTTCAGTATCTACGATT-3′)。引物序列详见酵母双杂交使用手册(Clontech,PT4084-1)。PCR 反应条件为:95℃预变性 5 min;95℃ 30 s,58℃ 30 s,72℃ 3 min,35个循环;72℃ 5 min;16℃保存。1%琼脂糖凝胶电泳检测扩增产物,统计重组率及插入片段长度。
1.2.3诱饵载体构建
参照无缝克隆试剂盒说明书,根据无毒基因PiAVR3b的核酸序列和pGBKT7载体酶切位点(EcoRⅠ和BamHⅠ),设计基因特异引物BD-PiAVR3b(表1),以致病疫霉菌株PIC99183的DNA为模板,利用DNA高保真聚合酶进行PCR扩增,将扩增产物回收纯化后与pGBKT7进行同源重组,转化大肠杆菌DH5α感受态细胞,筛选阳性克隆转化子,提取质粒,采用T7测序引物(表1)测序后获得诱饵载体pGBKT7-PiAVR3b。基因测序委托生工生物工程(上海)股份有限公司完成。

表1 酵母双杂交点对点验证试验引物1)Table 1 Primer sequences used in yeast two-hybrid peer to peer verification
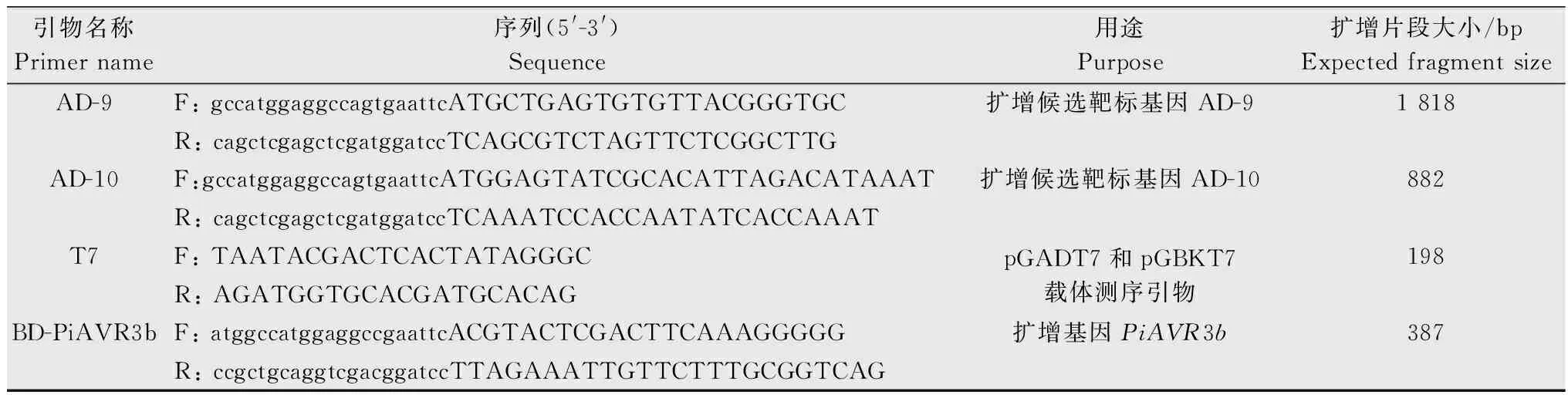
续表1 Table 1(Continued)
1.2.4诱饵载体自激活和毒性检测
挑取酵母菌株Y2HGold划线于YPDA 平板上,30℃恒温培养箱中倒置培养3 d左右,挑取单克隆酵母制备成感受态细胞,采用PEG/LiAc法将诱饵载体pGBKT7-PiAVR3b和pGBKT7空载体分别转化Y2HGold酵母感受态细胞,酵母菌液涂布于SD/-Trp营养缺陷培养基,置于30℃恒温培养箱培养,3~5 d后观察平板上酵母克隆的生长状态,若转化诱饵载体与转化空载体的克隆大小相近,生长数量近似,则证明pGBKT7-PiAVR3b对酵母菌株无毒性。将含有诱饵载体pGBKT7-PiAVR3b、空载体pGBKT7、53+T (阳性对照)、Lam+T (阴性对照)的Y2HGold酵母菌,分别接种于SD/-Trp/-His/-Ade/X-α-Gal平板上,30℃倒置培养3~5 d 后统计酵母生长情况。若pGBKT7-PiAVR3b在SD/-Trp/-His/-Ade/X-α-Gal平板上不能生长,则表明诱饵载体pGBKT7-PiAVR3b不存在自激活现象。若生长且变蓝色则说明诱饵载体存在自激活。
1.2.5PiAVR3b互作蛋白筛选
参照酵母双杂交使用手册(Clontech,PT4084-1),挑取直径2~3 mm含有诱饵载体pGBKT7-PiAVR3b的单克隆酵母菌株Y2HGold接种到50 mL SD/-Trp液体培养基中,30℃ 250~270 r/min过夜培养至OD600达到0.8;1 000 g离心浓缩去上清液,用4~5 mL SD/-Trp液体培养基重悬酵母菌并转移到2 L无菌三角瓶中;往三角瓶中加入1 mL含次级文库质粒的Y187酵母菌液和45 mL 2×YPDA液体培养基(含50 μg/mL Kan);30℃ 30~50 r/min共培养20~24 h。显微镜下观察杂交液是否出现米奇头或三叶草形状的结合子,有则继续下面的操作:1 000 g 离心10 min,用10 mL 0.5×YPDA(含有 50 μg/mL Kan) 重悬细胞,按每皿200 μL量涂布在SD/-Leu/-Trp/-His/X-α-Gal培养基上,培养5 d后,若出现蓝色酵母克隆,则说明可能存在与PiAVR3b互作的蛋白。挑选蓝色阳性克隆划线于SD/-Leu/-Trp/-His/-Ade/X-α-Gal培养基上扩大培养。同时,将阳性对照和阴性对照酵母菌也划线于SD/-Leu/-Trp/-His/-Ade/X-α-Gal培养基上培养。培养5 d后,提取正常生长酵母质粒,并以其质粒为模板,利用pGADT7载体测序引物(T7, 表1)进行PCR扩增,扩增产物送生工生物工程(上海)股份有限公司测序。测序结果在马铃薯基因组数据库(PGSC: http:∥solanaceae.plantbiology.msu.edu/)和NCBI数据库中进行BLAST比对分析。
1.2.6酵母双杂交点对点分析
为了验证酵母双杂交初筛的互作蛋白是否真的与PiAVR3b发生互作,用设计的10个候选互作基因特异引物AD系列(表1) 对‘合作88’的cDNA进行PCR扩增,以EcoRⅠ和BamHⅠ为酶切位点,将扩增产物通过无缝克隆试剂盒克隆到pGADT7载体中。将pGADT7-AD1至pGADT7-AD10分别与诱饵载体pGBKT7-PiAVR3b质粒共转化到Y2HGold酵母感受态细胞中。以53+T为阳性对照,Lam+T为阴性对照,涂布在SD/-Leu/-Trp和SD/-Leu/-Trp/-His/-Ade/X-α-Gal培养基上。30℃倒置培养3~5 d左右,若酵母正常生长且变蓝色则可判断初筛的互作蛋白与PiAVR3b发生互作。
2 结果与分析
2.1 致病疫霉诱导的马铃薯叶片cDNA文库构建和质量鉴定
将10 μL初级文库细菌原液稀释100倍后取50 μL稀释液涂布于LB平板(含50 mg/L Kan)上,次日统计,平板上共有单克隆1 600个,计算得出初级文库总库容量为1.28×107cfu/mL。随机挑取24个单克隆进行菌落PCR鉴定,24个单克隆均成功扩增,扩增片段平均长度在1 000 bp左右,说明初级文库中马铃薯cDNA插入率为100%(图1a和1b)。将10 μL次级文库细菌原液稀释100倍后进行次级文库库容量鉴定。取50 μL稀释液涂布LB平板(50 mg/L Amp)上,次日统计,平板上共有单克隆2 000个,计算得出次级文库总库容量为1.6×107cfu/mL。随机挑取24个单克隆进行菌落PCR鉴定,24个单克隆均成功扩增,扩增片段平均长度在1 000 bp左右,说明次级文库中马铃薯cDNA插入率为100%(图1c和1d)。结果表明,构建的马铃薯叶片cDNA文库容量足够大,可以满足筛选互作蛋白的文库要求。
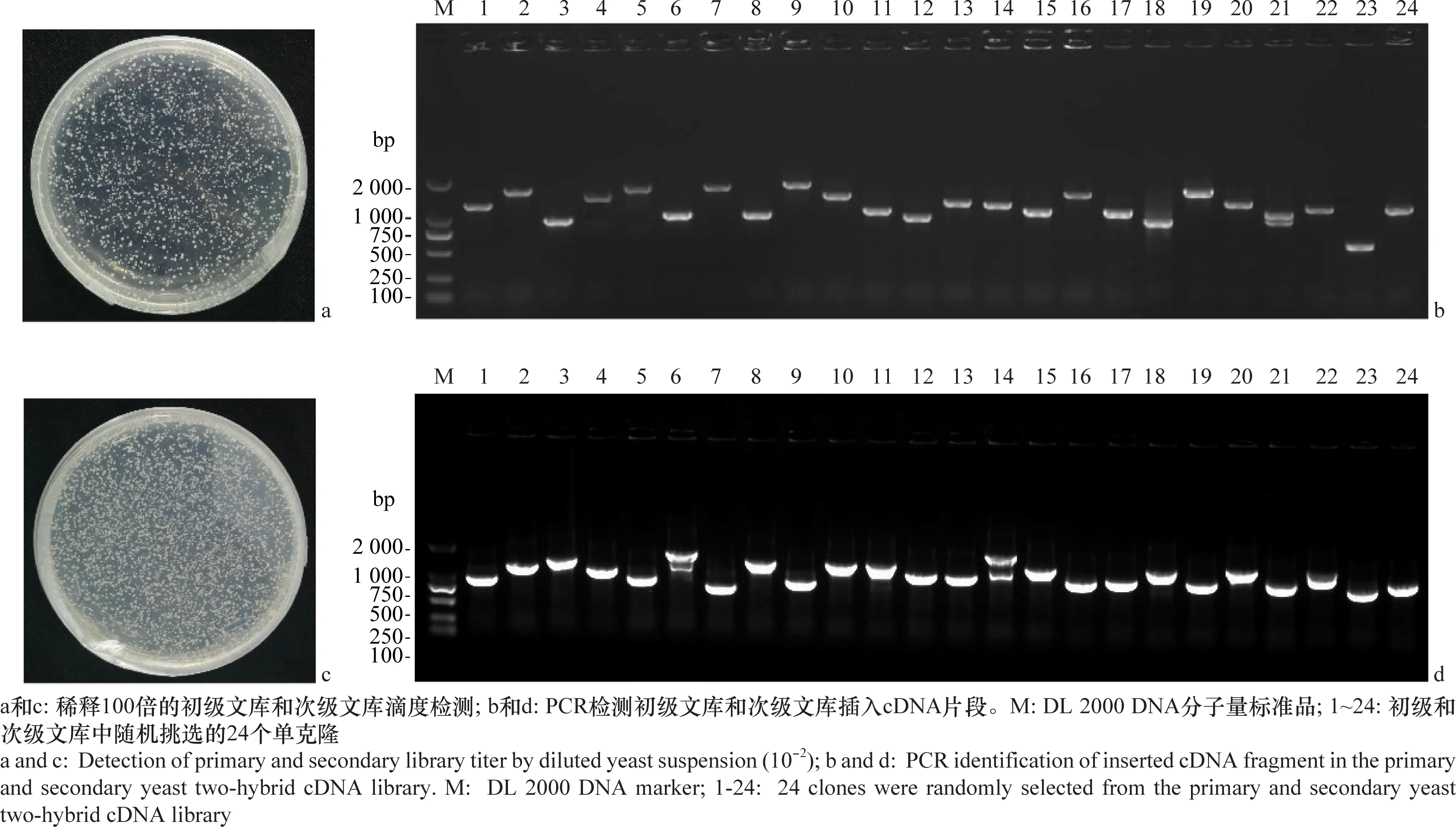
图1 致病疫霉诱导的马铃薯酵母双杂交初级、次级文库构建及质量检测Fig.1 Construction and evaluation of the primary and secondary yeast two-hybrid cDNA library in potato induced by Phytophthora infestans
2.2 诱饵载体pGBKT7-PiAVR3b构建
以致病疫霉菌株PIC99183的DNA为模板,BD-PiAVR3b为基因特异引物(表1),PCR扩增得到片段大小与预期目标片段大小一致(387 bp)(图2a)。利用胶回收试剂盒对扩增产物进行回收、纯化,通过同源重组方式将基因片段插入pGBKT7载体,转化大肠杆菌,菌落PCR筛选阳性克隆,提取阳性克隆的质粒,经酶切和测序验证确定诱饵载体pGBKT7-PiAVR3b构建成功(图2b)。
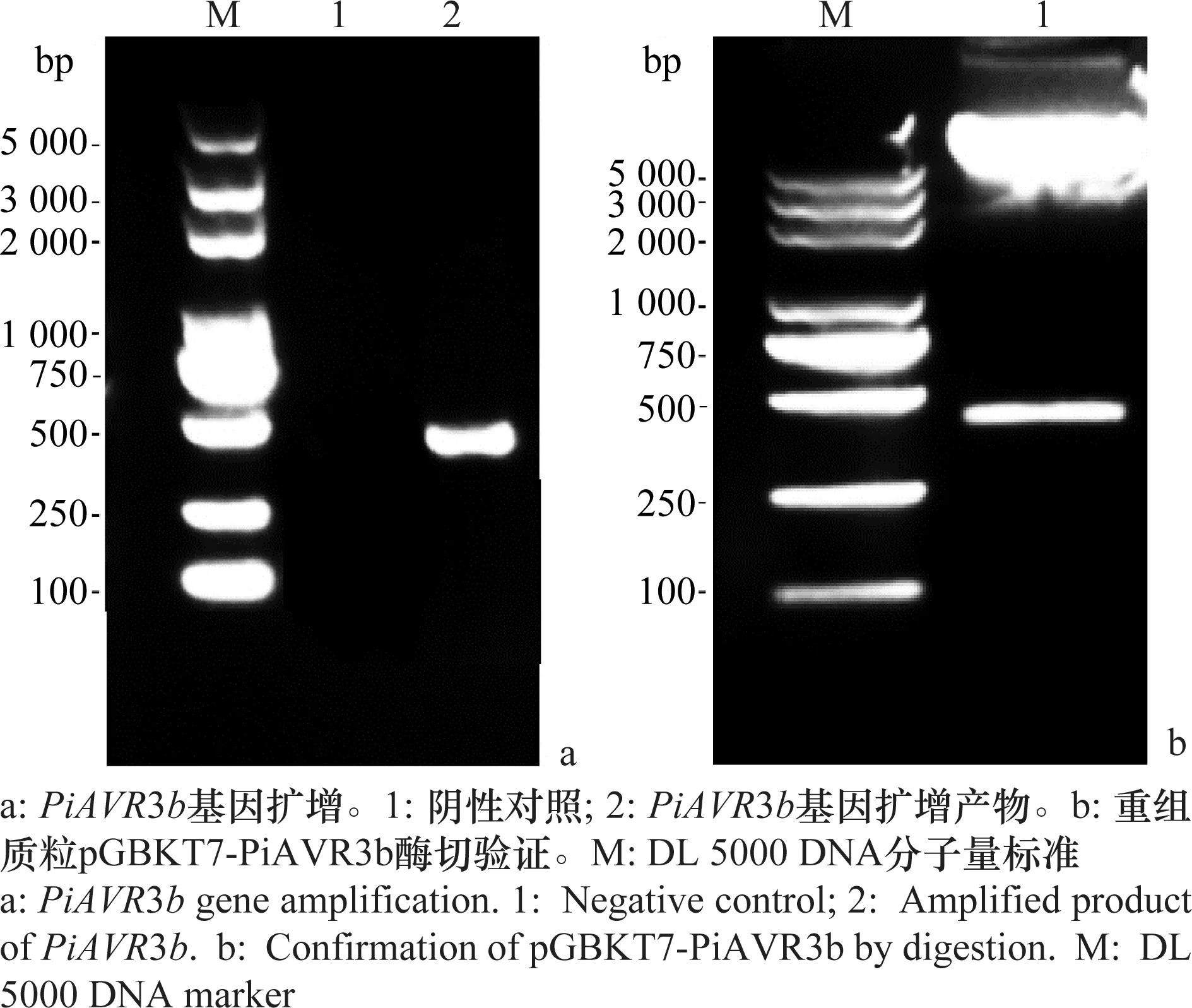
图2 pGBKT7-PiAVR3b诱饵载体构建Fig.2 Construction of bait vector pGBKT7-PiAVR3b
2.3 PiAVR3b自激活和毒性检测
以质粒组合53+T为阳性对照,质粒组合Lam+T为阴性对照,将构建好的pGBKT7-PiAVR3b和pGBKT7空载体按照Clontech酵母转化方案进行酵母细胞毒性和自激活验证。转化pGBKT7-PiAVR3b的酵母单克隆和转化pGBKT7空载体的克隆相比,大小和数量较一致,说明其对酵母细胞不产生毒性,可用于后续的马铃薯酵母双杂交文库筛选(图3a)。质粒pGBKT7-PiAVR3b和pGBKT7空载体均可在SD/-Trp培养基中正常生长,而不能在SD/-Trp/-His/-Ade/X-α-Gal培养基上生长,而阳性对照单克隆酵母可正常生长且显蓝色,阴性对照则不生长,表明构建的诱饵载体pGBKT7-PiAVR3b无自激活(图3b)。
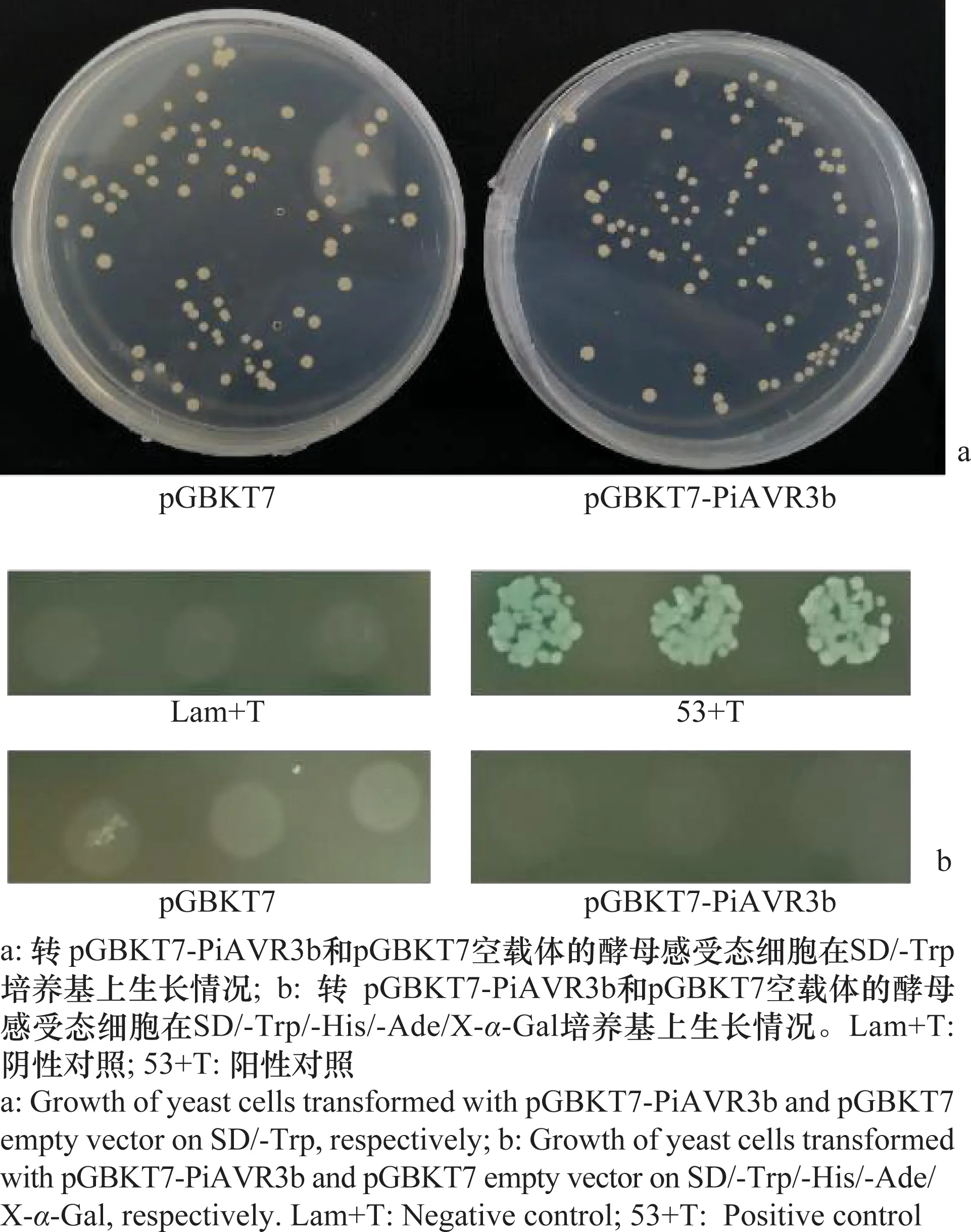
图3 pGBKT7-PiAVR3b诱饵载体自激活验证Fig.3 Validation of self-activation of bait vector pGBKT7-PiAVR3b
2.4 PiAVR3b互作蛋白的筛选
将含有诱饵载体pGBKT7-PiAVR3b的Y2HGold酵母菌株与含有经致病疫霉诱导的马铃薯酵母文库的Y187酵母菌株进行杂交,杂交24 h后涂布在SD/-Leu/-Trp/-His/X-α-Gal培养基上进行初筛,培养5 d后将初筛蓝色阳性克隆转移至SD/-Leu/-Trp/-His/-Ade/X-α-Gal平板上,经筛选后共获得28个阳性克隆(图4)。提取28个阳性酵母克隆的质粒,以质粒为模板,T7为引物进行PCR扩增,扩增产物直接进行纯化、回收、测序。测序序列经BLAST比对分析,去除相同序列的阳性克隆,初步获得10个候选靶标(表2)。
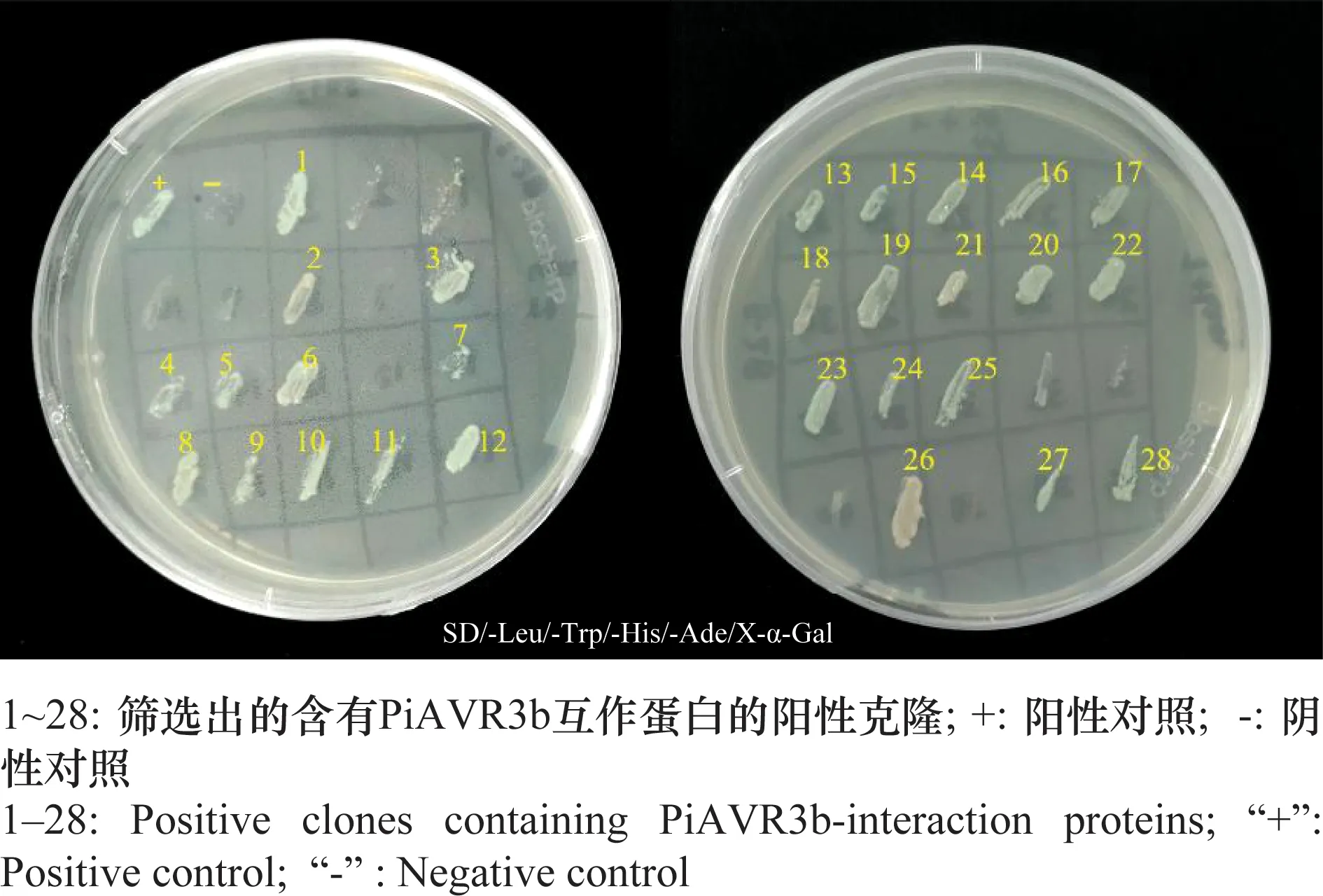
图4 无毒蛋白PiAVR3b互作蛋白的筛选Fig.4 Screening of candidate interaction protein for avirulence protein PiAVR3b

表2 无毒蛋白PiAVR3b的互作蛋白Table 2 Candidate interaction proteins of avirulence protein PiAVR3b
2.5 酵母双杂交点对点验证候选PiAVR3b靶标蛋白
为了进一步验证候选靶标与无毒蛋白PiAVR3b存在互作,将上述10个候选靶标基因的CDS序列分别重组连接到pGADT7载体上,将构建好的10个pGADT7-候选靶标质粒分别与pGBKT7-PiAVR3b共同转化Y2HGold酵母感受态细胞进行点对点验证,以53+T为阳性对照,Lam+T为阴性对照。结果发现在SD/-Leu/-Trp/-His/-Ade/X-α-Gal平板上只有4个候选靶标蛋白与PiAVR3b存在互作(图5),分别是马铃薯线粒体外膜孔蛋白(AD-6)、MYB-like蛋白(AD-7)、碳分解代谢抑制蛋白(AD-9)、肽基脯氨酰异构酶(AD-10)。
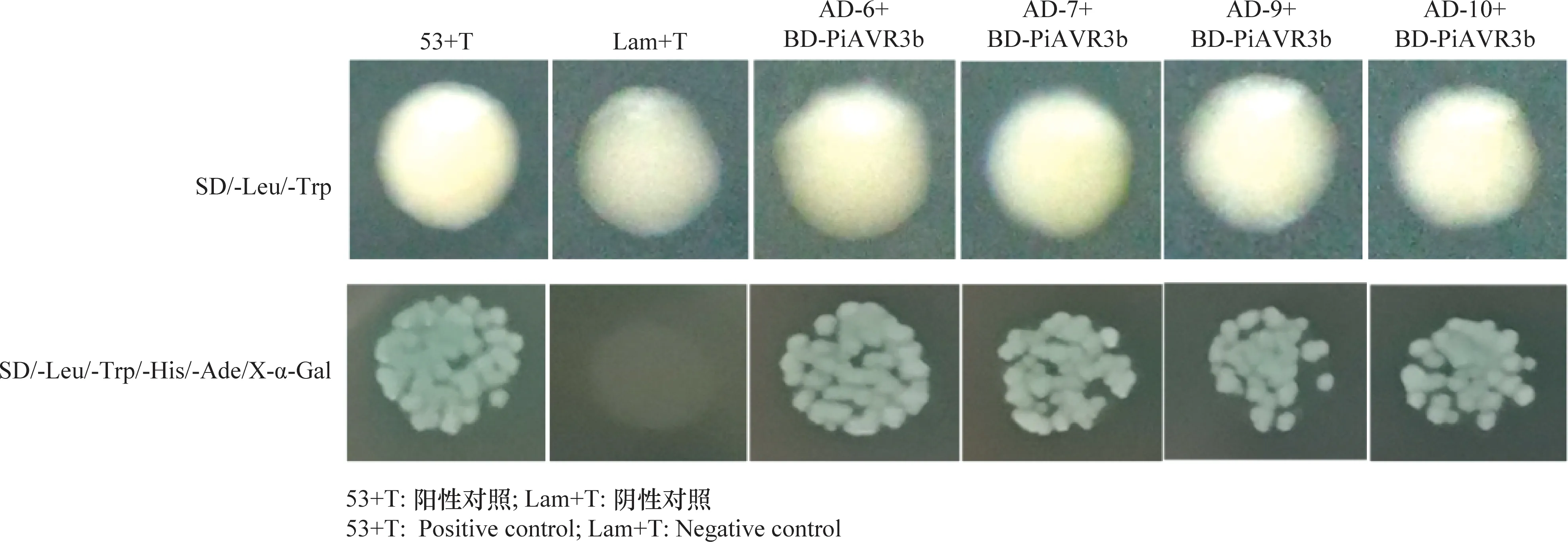
图5 酵母双杂交点对点验证无毒蛋白PiAVR3b 靶标蛋白Fig.5 Validation of the target proteins for avirulence protein PiAVR3b by using yeast two-hybrid peer to peer verification
3 讨论
高质量cDNA文库是酵母双杂交系统筛选互作蛋白的有力保证,而cDNA文库质量主要取决于RNA完整性、文库容量[16]。本研究构建的经致病疫霉诱导的马铃薯酵母双杂交次级文库库容量为1.6×107cfu/mL,插入片段平均在1 000 bp以上,重组插入率为100%,可以满足低丰度表达蛋白的互作筛选。对文库中10个互作蛋白的CDS序列分析结果显示,其中4个互作蛋白是全长CDS。上述结果说明构建的马铃薯酵母双杂交文库质量较好,能够用于致病疫霉效应蛋白的寄主靶标蛋白筛选。
RxLR效应蛋白通过调控植物转录因子、蛋白降解、泛素化、RNA 剪切等方面干扰植物免疫信号传导,从而促进致病疫霉侵染。然而,目前只发现了少数RxLR效应蛋白的寄主靶标[17]。筛选与鉴定效应蛋白的寄主互作靶标对进一步揭示效应子毒性功能和致病疫霉的致病机理至关重要。酵母双杂交系统是在真核酵母细胞内研究蛋白之间是否存在互作的一种方法,具有高灵敏度、广泛应用等特点。本研究构建了致病疫霉诱导的马铃薯酵母双杂交cDNA文库,并以无毒蛋白PiAVR3b为诱饵,筛选到4个候选靶标蛋白。经序列比对分析发现,这4个候选靶标蛋白分别是马铃薯MYB-like蛋白、线粒体外膜孔蛋白、碳分解代谢抑制蛋白、肽基脯氨酰异构酶。其中,碳分解代谢抑制蛋白在植物和微生物中功能保守,与植物免疫无关。推测其为假阳性互作蛋白。
MYB转录因子家族在植物生长发育的多个方面起着重要的作用,是植物中较大的转录因子家族之一[18]。木质素是植物细胞壁重要成分,在增强植物ETI免疫方面发挥着重要功能。研究发现拟南芥ArabidopsisthalianaMYB15突变体细胞壁中木质素含量降低,接种丁香假单胞菌Pseudomonassyringae无毒菌株后抗性下降,有病斑形成,而对照野生型则表现明显的过敏反应[19]。另外,拟南芥MYB20、MYB42、MYB43和MYB85转录因子均有促进次生细胞壁中的木质素和苯丙氨酸生物合成功能[20]。植物细胞表皮蜡质对抵抗生物胁迫具有重要功能。苹果MaluspumilaMdMYB30通过调控蜡质合成基因MdKCS1表达促使细胞表皮蜡质合成,从而增加对苹果轮纹病的抗性[21]。苹果MdMYB73通过调控体内水杨酸合成进而增强其对溃疡病的抗性[22]。超量表达番茄LycopersiconesculentumMYB49可以显著增强番茄植株对致病疫霉的抗性以及对干旱、盐胁迫等的耐受性[23]。另外,MYB转录因子在调控植物黄酮醇、花青素等次生代谢产物合成方面也发挥着重要作用[24]。课题组前期分析马铃薯MYB转录因子家族共有217个基因,其中1R-MYB类有90个,R2R3-MYB类有124个,R1R2R3-MYB类有3个[24]。本研究筛选获得的候选靶标蛋白MYB-like属于1R-MYB类,推测PiAVR3b靶向调控该转录因子影响寄主木质素,蜡质或水杨酸等合成,从而导致马铃薯抗病性降低。
线粒体外膜孔蛋白负责线粒体与细胞质之间的信息传递,对细胞功能的正常发挥具有重要作用。在植物受到病原菌侵染时,部分线粒体外膜孔蛋白被诱导表达,促使活性氧从线粒体向细胞质释放从而增强植物抗病免疫[25]。研究发现线粒体外膜孔蛋白VpVDAC3通过与病程相关蛋白VpPR10.1互作增强葡萄对霜霉病菌的抗性[26]。拟南芥受丁香假单胞菌侵染时,体内4个线粒体外膜孔蛋白均上调表达[27]。通过蛋白质组学技术,研究人员发现线粒体外膜孔蛋白在油菜抗黑腐病品种中表达丰度明显高于感病品种[28]。候选靶标线粒体外膜孔蛋白有可能参与马铃薯抗晚疫病,同时与PiAVR3b发生互作,两者之间如何调控马铃薯对晚疫病的抗性有待深入研究。
肽基脯氨酰异构酶广泛参与植物响应生物与非生物胁迫反应[29]。研究发现丁香假单胞菌侵染拟南芥时,肽基脯氨酰异构酶类AtCYP19-1和AtCYP57基因被诱导表达。当抑制AtCYP19-1和AtCYP57基因表达时,植株表现感病,超量表达则增强植株抗病性。进一步研究发现,AtCYP19-1和AtCYP57超量表达植株促进类活性氧爆发和胼胝质积累从而增强植株抗病性[30-31]。肽基脯氨酰异构酶FKBP15在拟南芥内质网胁迫感知和介导植物免疫方面发挥重要功能,研究发现辣椒疫霉Phytophthoracapsici无毒蛋白PcAvr3a12可以抑制FKBP15酶活从而促进辣椒疫霉侵染拟南芥[32]。另外,肽基脯氨酰异构酶类AtCYP20-3以茉莉酸合成途径中对12-氧-植物二烯酸的受体形式调控拟南芥对腐生真菌的抗性[33]。本研究中,候选靶标肽基脯氨酰异构酶是否影响致病疫霉侵染及PiAVR3b是否依赖其发挥毒性有待进一步研究。
4个候选靶标蛋白均与PiAVR3b互作,一方面可能由于PiAVR3b通过靶向寄主不同蛋白来调控植物免疫,类似PiAVR-blb2[34-35]。因此需要对4个候选靶标是否参与寄主抗病免疫反应进行鉴定。另一方面由于酵母双杂交技术存在一定的假阳性[17],需要进一步通过其他蛋白质互作技术对候选靶标与PiAVR3b的互作真实性进行鉴定,例如蛋白质免疫共沉淀、双分子荧光互补、蛋白体外pull-down技术等。通过上述两个方面进行PiAVR3b靶标蛋白鉴定,将为进一步揭示PiAVR3b的作用机制提供依据。
综上,本研究构建了经致病疫霉诱导的马铃薯酵母双杂交cDNA文库,文库具有较高转化效率和重组效率,并以致病疫霉无毒蛋白PiAVR3b为诱饵,经酵母双杂交筛选到4个候选PiAVR3b靶标蛋白,为后续研究PiAVR3b如何调控植物免疫及致病疫霉与马铃薯互作奠定基础。
猜你喜欢
湘潮(上半月)(2022年7期)2022-12-06
今日农业(2022年4期)2022-11-16
猪业科学(2021年3期)2021-05-21
军民两用技术与产品(2021年10期)2021-03-16
湖南工业大学学报(2020年6期)2020-11-27
幽默大师(2020年10期)2020-11-10
中国畜牧业(2019年22期)2019-12-30
中华诗词(2019年1期)2019-11-14
世界农药(2019年3期)2019-09-10
名人传记·财富人物(2017年9期)2017-11-02
