
苯硫基烯酮与不活泼烯烃的分子内[2+2]环加成反应
2022-08-04 04:03邓威力李有桂
合肥工业大学学报(自然科学版) 2022年7期
吴 祥, 何 浩, 汪 涛, 邓威力, 李有桂
(1.合肥工业大学 化学与化工学院,安徽 合肥 230009; 2.合肥工业大学 先进催化材料与反应工程安徽省重点实验室,安徽 合肥 230009; 3.福建师范大学 生命科学学院,福建 福州 350117)
0 引 言
环丁烷衍生物是一类重要的有机合成中间体,易于开环和扩环的特点使其在天然产物的合成中得到了广泛的应用[1-2]。烯酮与烯烃环加成反应是合成环丁酮衍生物的常用方法[3]。其中,烯酮与烯烃的分子内[2+2]环加成反应生成具有高度区域和立体控制的且具有多种合成用途的多环环丁酮化合物[4],并应用于天然产物的全合成[5]。
然而,近年来,烯酮与烯烃的分子内[2+2]环加成反应的发展受到了连接简单烷基烯酮的链长和烯烃反应活性的影响。由于系链长度的限制,仅当烯酮和烯烃部分之间的系链是3个碳原子时,才能给出最好的结果。对于较长的系链,分子内环加成反应是相当罕见的[6],只有用烯酮亚胺盐和烷氧基烯酮以及在具有构象限制的系链的情况下才能实现[7-8]。简单烷基烯酮的反应活性有限,环加成的产率低。例如,文献[9]通过用三乙胺处理酰氯1制备的烯酮仅提供3%产率的环加成物2,如图1a所示。
然而,在酰基氯1的羰基的α位引入氯原子后,酰基氯3以68%的产率得到环加成物4,如图1b所示。由于烯酮片段在环加成中作为亲电组分,因此烯酮上的其他吸电子取代基(如氧或硫)可以增强反应活性。氯原子[7]和氧原子[8,10]作为取代基已被应用于分子内烯酮的环加成反应中。然而,尽管已有报道苯硫基烯酮或烷基苯硫基烯酮与活化烯烃的分子间[2+2]环加成反应以较高的产率得到环加成产物[11],但目前有关硫代烯酮与烯烃的分子内[2+2]环加成反应的报道并不多。同时,环加成物的还原脱硫可以在温和的条件下进行,并且硫代烯酮可以替代现有的氯代烯酮。因此,本文开展了烷基苯硫基烯酮与不活泼烯烃的分子内[2+2]环加成反应来构建多环环丁酮的研究。

图1 烯酮分子内[2+2]环加成反应
1 实验部分
1.1 合成α-苯硫基羧酸的通用方法
在低温(-10 ℃)以及氮气保护条件下,正丁基锂(3.8 mL、6.0 mmol,1.2当量)加入到二异丙胺(0.78 mL、5.5 mmol,1.1当量)的THF(10 mL)溶液中。搅拌0.5 h后,在-78 ℃下,羧酸酯(5 mmol,1当量)的THF(2 mL)溶液加入到反应体系中。反应1 h后,二苯二硫醚(1.53 g、7.0 mmol,1.4当量) 的THF (2 mL)溶液加入到反应体系中,室温反应2 h。薄层色谱法(thin-layer chromatography,TLC)检测到反应结束后,加入水淬灭反应,并用乙醚萃取3次。依次用0.1 mol/L盐酸,饱和碳酸氢钠溶液和饱和食盐水洗涤有机相,无水硫酸钠干燥。抽滤,浓缩有机相,柱层析纯化 (V(石油醚)∶V(乙酸乙酯)=30∶1),得到无色油状液体,即为α-苯硫基羧酸酯。
LiOH溶液(4%,12 mL)缓慢加入到α-苯硫基羧酸酯(1.67 g、4 mmol,1当量) 的THF(36 mL)和MeOH(12 mL)的混合溶液中。反应3 h,TLC检测到反应结束后,加入1 mol/L盐酸淬灭反应,并调pH值至3~4。用乙酸乙酯萃取,饱和食盐水洗涤有机相,无水硫酸钠干燥。抽滤,浓缩有机相,柱层析纯化(V(石油醚)∶V(乙酸乙酯)=10∶1),即可得到α-苯硫基羧酸。
1.2 [2+2]环加成反应的通用方法
氮气保护条件下,三乙胺(8当量)和Mukaiyama 试剂(4当量)溶在20 mL乙腈中,加热至80 ℃,α-苯硫基羧酸(1.5 mmol)溶在乙腈(30 mL)中滴加到反应体系中,4 h滴加完毕,继续反应4 h。TLC检测到反应结束后,冷却至室温,旋去溶剂,加入1 mol/L盐酸,用乙醚萃取,饱和食盐水洗涤有机相,无水硫酸钠干燥。抽滤,旋干溶剂,柱层析得到相应的环丁酮或还原二聚体以及烯胺酮产物。
2 结果与讨论
作为不饱和烷基苯硫基烯酮的前体,相应的羧酸通过两步反应很容易制备得到。首先,通过与二异丙基胺基锂反应将不饱和酯转化为烯醇锂,然后在-78 ℃下添加二苯二硫醚淬灭,高产率得到α-苯基硫代产物(80%~90%);随后在THF/MeOH/H2O的混合溶剂中用LiOH水解α-苯硫醚得到定量产率的羧酸5,具体反应方程式为:

得到不饱和α-苯硫基羧酸后,制备烷基苯硫基烯酮。文献[11]中,硫取代的烯酮可以分别通过由三乙胺促进的酰基氯的脱卤化氢、Fischer铬卡宾配合物的光化学重排或通过铑催化的硫杂Wolff重排等手段合成得到。
本文最初通过酰氯的脱氯化氢生成烷基苯硫基烯酮。α-苯硫基羧酸5a与草酰氯在苯中反应,得到酰氯6。将酰氯6缓慢加入到三乙胺的二氯甲烷溶液中,并继续反应4 h。然而,反应液在用稀盐酸处理后,不饱和苯硫基烯酮以低产率(5.7%)得到所需的合物7a,并大量回收原料,如图2所示。即使在更长时间、更高稀释度和更高温度条件下,该反应也无法获得更好的结果。

图2 酰氯脱氯化氢生成α-苯硫基乙烯酮
因此选用另一种方法制备烷基苯硫基烯酮。2-氯-1-甲基吡啶碘盐(Mukaiyama试剂8)已被广泛用于活化羧酸以提供酯、酰胺或内酯[12],并且还可以实现羧酸原位脱水为烯酮,随后用于与烯烃、联烯[13]和醛类[14]的[2+2]环加成反应[15-16]。根据Romo的反应条件[15],在4 h内(用注射泵)将5a加入Mukaiyama试剂8(4.0当量)和Et3N(8.0当量)的混合溶液中,然后再搅拌4 h,以70%的收率得到α-苯硫环丁酮7a。反应方程式如下:

本文对其他条件(如溶剂、温度和反应时间)进行了优化,相关数据见表1所列,其中序号2的条件为最优。

表1 分子内[2+2]环加成反应的优化条件
在最优的条件下,本文还考察一系列不饱和α-苯硫基羧酸发生分子内[2+2]环加成反应的情况,结果见表2所列。从表2可看出,由5a衍生的烯酮得到70%产率的7a,而非苯硫基羧酸5b衍生的烯酮没有得到环加成产物。

表2 不饱和α-苯硫基羧酸分子内[2+2]环加成反应产物
##表示底物中不含苯硫基,相应位置被甲硫基取代。
对比图1a的结果可以看出,苯硫基的引入可以提高烯酮的反应活性,不饱和苯硫基烯酮的分子内环加成反应比不含苯硫基的烯酮更容易发生。从5c衍生的具有4个碳原子链的烯酮,以62%产率得到7c。与二取代末端烯烃5c相比,单取代烯烃5d和1,2,2-三取代烯烃5e、5f没有得到环加成产物,但得到还原性二聚体9d~9f和烯胺酮10d~10f。因此,具有5c取代模式的烯烃比具有5d~5f取代模式的烯烃相对更具反应性。含有五原子系链的5g不能发生分子内环加成,但以64%的产率转化为还原性二聚体9g,以31%的产率转化为烯胺酮10g,具体反应方程式如下:

不含苯硫基的酸5h在标准条件下没有发生相应的反应,而α-苯硫基羧酸5i以70%产率得到三环加成产物7i,其非对映体的比例约为2.5∶1。表2中,甲硫基取代酸5j也可以得到三环加合物7j,产率为57%,低于苯硫基取代酸5i的产率,苯硫基对环加成反应很重要,且苯硫基取代的烯酮的反应活性高于甲硫基取代的烯酮。酸5k除了得到30%产率还原性二聚体9k和12%产率的烯胺酮10k之外,仅得到20%产率的三环加合物7k。底物5l~5n中位于六元环的不同取代模式的双键不发生反应,但产生还原性二聚体和烯胺酮。
修饰过的羧酸5o以60%的产率得到三环加合物7o,并通过X射线衍射(X-ray diffraction,XRD)证实其结构,化合物7o可进一步转化为桉烷萜类等天然产物,反应方程式如图3所示。

图3 三环骨架的构建
基于上述结果可以推断,尽管底物5c、5i~5k含有不利的四原子系链和不活泼的烯烃,但它们仍然以20%~70%的产率提供环加成产物。这一现象是由于苯硫基取代的烯酮的高反应活性和六元碳环的限制引起的。但是,具有4个原子的酸5d~5g、5l~5n也转化为苯硫基取代的烯酮,不能得到任何环加成产物,这是由于具有取代模式的烯烃更不活泼。根据前线分子轨道(frontife molecular orbital,FMO)理论,烯烃和烯酮之间的初始相互作用须形成八元过渡态,但对于底物5l~5n很难形成桥联的八元环中间体。
当烯烃不反应时,得到还原性二聚体9和烯胺酮10,其为本文首次发现,通过核磁共振、傅里叶变换红外光谱和高分辨质谱可以确认该类化合物的结构。本文推测形成这2种化合物的可能反应机理如图4所示。α-苯硫基羧酸用2-氯-1-甲基吡啶碘盐处理后,转化为苯硫基取代的烯酮11。二聚体12由苯硫基取代的烯酮11二聚形成,随后被Et3N进攻得到α-酰氧基三乙基铵13。经过E2消除,α-酰氧基三乙基铵13转化为α-酰氧基乙烯基阴离子14和亚胺正离子15。乙烯基阴离子14质子化后得到还原性二聚体9。亚胺正离子15烯胺化得到烯胺16,其进一步攻击烯酮11得到亚胺正离子17。中间体17再次进行烯胺化,得到烯胺基酮10。
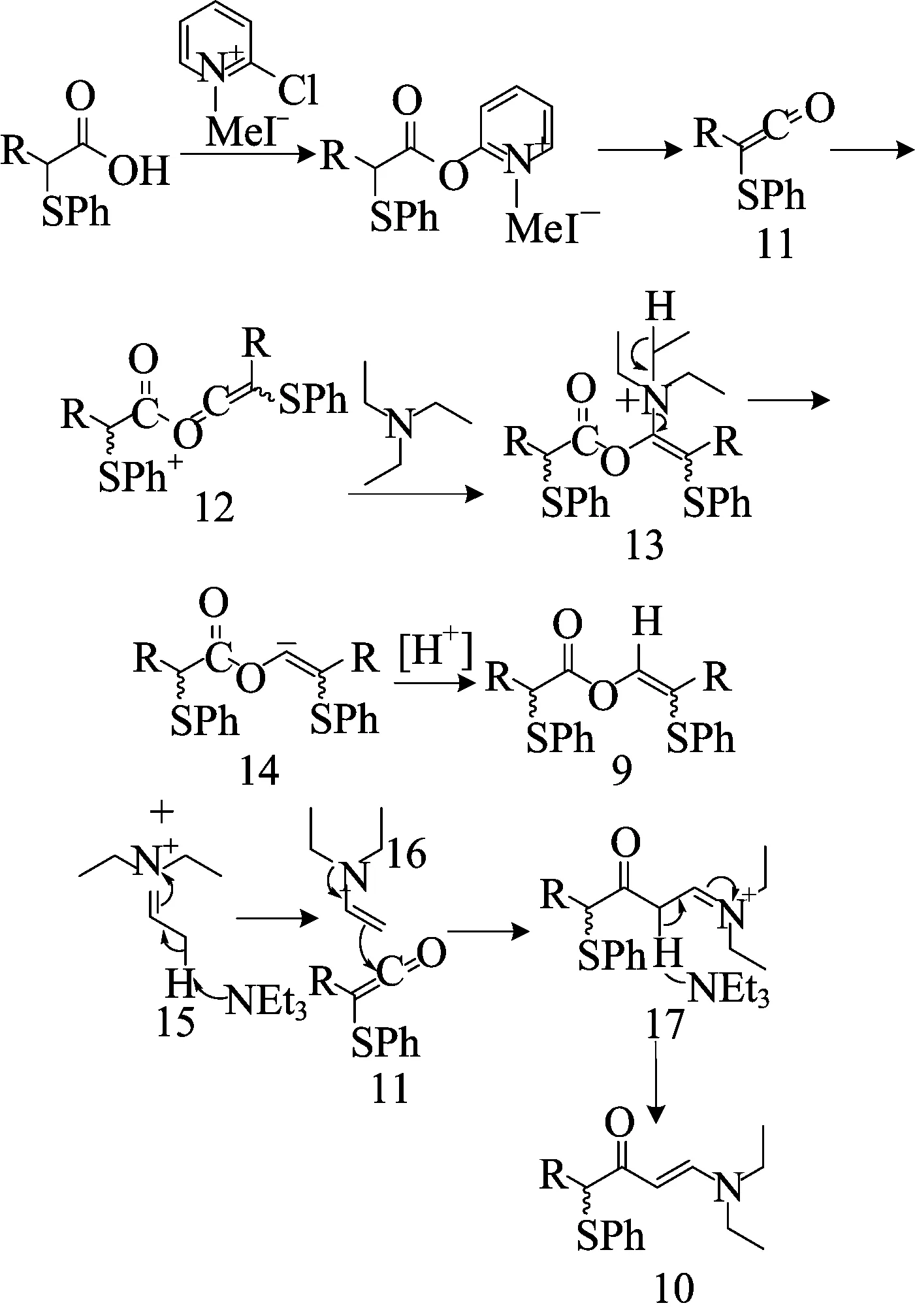
图4 产物9和产物10生成的可能机理
3 结 论
本文提出由α-苯硫基羧酸与不活泼的烯烃经苯硫基烯酮中间体发生分子内[2+2]环加成反应的新方法,该方法提供了一条合成双环丁酮的通用路线。此外,当烯酮和烯烃之间的4个碳原子受到六元碳环的限制时,也可以得到三环环丁酮,该方法可应用与天然产物及其衍生物的全合成中。同时,对于不能发生环加成反应的底物,得到还原性二聚体和烯胺酮产物,并提出了可能的反应机理。
猜你喜欢
炼油与化工(2022年4期)2022-10-10
中国典型病例大全(2022年12期)2022-05-13
科学家(2022年4期)2022-05-10
浙江临床医学(2022年1期)2022-03-02
家庭医学·下半月(2021年7期)2021-08-19
现代仪器与医疗(2021年2期)2021-07-21
今日农药(2017年10期)2017-11-14
进出口经理人(2017年12期)2017-10-23
山东工业技术(2017年12期)2017-07-06
