
自支撑阴极用于宽pH域原位产H2O2及其耦合UV降解卡马西平
2022-07-28 01:57曹建新徐昊林冯春华
环境科学研究 2022年7期
曹建新,张 露,严 樟,徐昊林,冯春华
华南理工大学环境与能源学院,工业聚集区污染控制与生态修复教育部重点试验室,广东 广州 510006
过氧化氢(H2O2)作为基础化工物质,已被广泛应用于污染治理[1-6]、能源应用[7-8]等领域. 现阶段,工业生产H2O2主要依赖于蒽醌工艺,该过程需要使用有毒有机溶剂,容易产生二次污染. 另外,高浓度H2O2的长距离运输及存储存在较大挑战[9]. 因此,亟需开发安全、高效、原位合成H2O2的新技术. 近年来,大量研究者致力于电催化合成H2O2的研究[10-13],其原理为通过阴极二电子氧还原反应(2e-ORR)将O2原位还原为H2O2〔见式(1)〕,其能够有效避免H2O2存储及运输面临的安全问题,且无有毒副产物生成,是理想的H2O2合成新技术. 因H2O2活化产生的·OH〔见式(2)(3)〕具有高达2.7 V的氧化电位[14],能够氧化降解绝大多数有机污染物[15-16],电催化合成H2O2已应用于电芬顿及UV/H2O2等高级氧化体系. 电催化合成H2O2的核心是阴极材料的开发,目前常用的阴极材料主要有贵金属、杂原子掺杂碳基材料、单原子催化剂及金属化合物等[11],其中杂原子掺杂碳基材料具有造价低、高导电性、高化学稳定性等优势,十分适合用于电催化产H2O2[17-18].

近期有关电催化产H2O2的碳基阴极材料研究主要集中于酸性及弱酸性条件下2e-ORR性能评估方面[19-22],对宽pH域内电催化产H2O2的研究却鲜见报道. 针对上述问题,该研究旨在开发出一种可在宽pH域内能有效合成H2O2的杂原子掺杂碳基阴极材料. 最近,Al-Zoubi等[23]以Cd为牺牲金属,于750 ℃下低温热解,制备铁单原子为活性位点的碳基材料Fe-C-N750,由于单原子Fe的掺杂,其展现出优异的4e-ORR性能. 该研究借鉴其低温热解思路,以Cd为牺牲金属,以碳布为基底,成功于750 ℃的煅烧温度下合成不含Fe的自支撑碳基杂原子掺杂材料(750/N@C-CC). 由于没有单原子Fe介导4e-ORR的过程,保证了750/N@C-CC具备较好的2e-ORR性能,可有效合成H2O2;同时自支撑结构能够有效保障材料的电子传递性能[24].
该研究通过煅烧法合成一种自支撑碳基杂原子掺杂阴极材料,其在宽pH域均能有效原位电催化产H2O2,并考察煅烧温度对材料性能的影响、探讨H2O2合成机理;同时以CBZ为目标污染物,研究UV活化H2O2体系在不同pH条件下对CBZ的降解性能,结合自由基掩蔽试验与电子顺磁共振(EPR)探针试验考察污染物降解机理. 该研究开发了具有宽pH工作域的电催化合成H2O2阴极材料,通过UV耦合电催化产H2O2的方式,无需外加铁源直接生成氧化活性物种降解污染物,避免了传统芬顿过程中铁泥产生和pH局限,具有良好的应用前景.
1 材料与方法
1.1 药剂及材料
四水合硝酸镉〔Cd(NO3)2·4H2O〕、N,N-二甲基乙醇胺(DABCO)、对苯二甲酸(PTA)、二甲基甲酰胺(DMF)、卡马西平(CBZ)、草酸钛钾和二甲基吡咯烷酮(DMPO)均购于阿拉丁生物科技有限公司,无水硫酸钠、氢氧化钠、硫酸、无水乙醇、甲醇、乙腈和叔丁醇均购于广州化学试剂厂,以上试剂除乙腈和甲醇为色谱纯,其余均为分析纯;碳布购于苏州晟尔诺科技有限公司;钌铱电极(MMO)购于陕西昌立特种金属有限公司;紫外光源(275 nm,20 W)购于珠海市天辉电子有限公司.
1.2 电极制备
1.2.1碳布预处理
将碳布裁剪成3 cm×3 cm大小,而后依次于无水乙醇和去离子水中超声30 min,紧接着用去离子水冲洗干净,并于80 ℃真空条件下干燥过夜.
1.2.2电极制备
分别取0.5 g Cd(NO3)2·4H2O,0.55 g DABCO,0.8 g PTA溶于35 mL DMF中,而后于150 ℃油浴中加热搅拌2 h,待冷却至室温后即形成白色前驱体悬浊液.将预处理过碳布浸泡于悬浊液中静置5 h,随后将浸泡好的碳布于80 ℃真空下干燥过夜. 最后,将干燥好的碳布置于管式炉中,以5 ℃/min的升温速率,于750 ℃氩气氛围下保持30 min,待煅烧完冷却至室温后,即得到750/N@C-CC. 对照组电极的煅烧温度为550和950 ℃(550/N@C-CC和950/N@C-CC),其他合成条件不变.
1.3 阴极材料表征
采用场发射扫描电子显微镜(FESEM, Melin,Germany)对纯碳布及负载氮掺杂碳材料碳布的形貌进行表征;通过X射线衍射仪(XRD, X′Pert PRO MRD, the Netherlands)分析材料晶体结构;利用X射线光电子能谱(XPS, ESCALAB 250Xi, America)分析材料元素组成,拉曼光谱(Raman, Lab RAM Aramis,France)分析材料石墨化程度及缺陷程度;傅里叶变换红外吸收光谱仪(FT-IR, VERTEX 33, Bruker)用以分析阴极材料上的含氧官能团;通过电子顺磁共振波谱仪(EPR, EMX-10/12, Bruker, Karlsruhe,Germany)分析UV/H2O2体系产生的自由基种类.
1.4 电化学测试
利用CHI 660E电化学工作站构建三电极体系,其中工作电极为氮掺杂碳材料,辅助电极为饱和甘汞电极,对电极为铂网电极. LSV测试条件为扫描电位-0.7~0.2 V (vs. SCE)、扫描速率50 mV/s、溶液pH=6.通过分析阴极材料的极化曲线和EIS阻抗图判断其ORR性能.
1.5 样品测定方法
H2O2浓度测定采用草酸钛钾分光光度法[25],CBZ浓度采用高效液相色谱测定,配有BEH C18色谱柱,其流动相为乙腈和水(体积比为60∶40),进样体积为20 μL,流速为0.8 mL/min,检测波长为285 nm,柱温为30 ℃.
1.6 电化学试验
1.6.1H2O2产率试验
利用直流电源构建两电极体系:阴极为碳布负载氮掺杂碳材料(3 cm×3 cm);阳极为MMO (3 cm×3 cm);电流大小为10~30 mA;初始pH为3~10;电解质为0.05 mol/L Na2SO4;反应体积100 mL;反应时长1 h.
1.6.2CBZ降解试验
利用直流电源构建两电极体系:阴极为碳布负载氮掺杂碳材料(3 cm×3 cm);阳极为MMO (3 cm×3 cm);电流大小为20 mA;初始pH=3、6、10;电解质为0.05 mol/L Na2SO4;紫外光源为UV275;反应体积100 mL;CBZ浓度为10 μmol/L;反应时长30 min.
2 结果与讨论
2.1 阴极材料形貌及组分分析
为研究氮掺杂材料在碳布上的负载情况,利用SEM对碳布表面形貌进行表征. 如图1所示,碳布由众多碳纤维(CC)均匀编织而成,具有良好的机械强度、化学稳定性和导电性,同时其良好的三维编织结构也为杂原子掺杂材料负载提供了充足的位点[26]. 经浸泡后的碳布纤维表面均匀负载了长方体状前驱体,表明前驱体的成功合成及负载. 经过高温煅烧后,前驱体转化为多孔膜状材料,且与碳布结合紧密;同时,碳布整体结构及形貌在煅烧前后无明显改变,说明该材料合成过程不会对碳布基底产生影响. 通过分析750/N@C-CC的XRD图谱(见图2),发现其在25°和44°附近有两个明显的衍射峰,其分别代表了碳的(002)和(100)晶面[27].

图1 原始碳布及负载前驱体碳布煅烧前后SEM图Fig.1 SEM images of as-received carbon cloth and carbon cloth loaded with precursor before and after calcination

图2 750/N@C-CC XRD图谱Fig.2 XRD spectrum of 750/N@C-CC
为明晰阴极材料电催化合成H2O2的主要活性位点,探讨了不同煅烧温度对材料元素组成的影响. 借助XPS图谱对550、750、950 ℃ 3个温度煅烧后的材料(即550/N@C-CC、750/N@C-CC、950/N@C-CC)组成进行分析. 如表1所示,碳材料中所含主要元素为C、N、O、Cd. 前驱体中含有1.99%的Cd,550 ℃煅烧温度下Cd含量降至0.47%,750及950 ℃煅烧温度下Cd几乎已挥发完全,侧面印证造孔较为充分.温度升高使得碳材料中N元素相对含量逐渐降低,其中550/N@C-CC、750/N@C-CC、950/N@C-CC三个样品中N含量分别为14.17%、8.89%、6.61%. 可见,煅烧温度对Cd、N含量影响较大. 此外,笔者对550/N@C-CC、750/N@C-CC、950/N@C-CC中N元素进行分峰拟合,如图3所示,N1s图谱被拟合为吡啶氮(Pyridinic-N)、吡咯氮(Pyrrolic-N)和石墨氮(Graphitic-N) 3个部分. 其中,398.6 eV处为吡啶氮,399.8 eV处为吡咯氮,400.9 eV处为石墨氮[28]. 拟合结果显示,温度的升高对3种掺杂氮含量产生了较大影响,随煅烧温度的升高,吡啶氮含量从550 ℃时的6.38%依次降至750 ℃时的4.18%和950 ℃时的2.96%,但总氮中吡啶氮的相对含量分别变为45.08%、47.01%和44.83%,其中,750 ℃时吡啶氮的相对含量最高. 550 ℃时吡咯氮的含量为3.23%,当煅烧温度升至750 ℃以上时,吡咯氮全部转化或从碳材料中剥离;而550、750、950 ℃时石墨氮的含量分别为4.55%、4.71%和3.65%,其中750 ℃时拥有最高的石墨氮含量. 据报道显示,石墨氮位点能够向与其π共轭的C原子提供电子,进而诱导电荷重新分布,以增强其相邻α-C原子的氧吸附能力[29];吡啶氮位点能有效提高ORR起始电位[30-31],从而促进H2O2的合成.

图3 前驱体及不同煅烧温度后的XPS全谱、XPS N1s、氮含量及种类分布分析Fig.3 XPS full spectra, XPS N1s spectra, and N content (atomic) and N species distribution of precursor and X/N@C-CC

表1 碳布负载前驱体及不同温度煅烧后C、N、O、Cd的相对含量Table 1 Relative composition of C, N (Pyridinic-N, Pyrrolic-N and Graphitic-N), O, and Cd atoms in precursor and X/N@C-CC
在中碱性条件下,含氧官能团种类(如C-O、C=O和-COOH)能够作为关键活性位点,降低2e-ORR过电势并提高H2O2选择性[32-33]. 因此,碳材料中含氧官能团对电催化产H2O2有重要意义. 如图4及表1所示,通过XPS拟合分析发现,550 ℃后煅烧温度的升高对材料中氧种类及含量无太大影响,在3个煅烧温度下均分析出了C-O、C=O及-COOH官能团. 此外,还通过FTIR谱图研究煅烧温度对含氧官能团变化的影响. 如图4所示,其中1 600 cm-1处为C=C及-COOH,1 550 cm-1处为-COOH,1 300 cm-1处为C-C,1 120 cm-1处为C-O,760 cm-1及528 cm-1处均为C=O. FTIR光谱结果印证了XPS拟合结果,证明3种煅烧温度下均含有C-O、C=O及-COOH官能团. 另外,碳原子晶格不匹配导致的原子重排会引起碳材料内部拓扑缺陷产生,促进电子的重新分布,从而增强碳材料ORR活性[34]. 通过拉曼表征可知,1 343和1 586 cm-1处出现了两个明显特征峰即D峰和G峰,其中,D峰代表碳材料缺陷程度,G峰代表碳材料石墨化程度.ID/IG值越大,材料缺陷程度越高,ID/IG值越小,材料石墨化程度越高. 结果显示,相较于550/N@C-CC,750/N@C-CC和950/N@C-CC样品的缺陷略有增加.

图4 X/N@C-CC的XPS O1s、FTIR及拉曼图谱Fig.4 XPS O1s, FTIR, and Raman spectra of X/N@C-CC
2.2 电化学表征
为考察不同煅烧温度对阴极材料ORR活性的影响,研究了其在溶解氧饱和条件下LSV曲线和电化学阻抗(见图5). 750/N@C-CC拥有最高的ORR起始电位,这是因为其拥有最高的吡啶氮相对含量(XPS分析结果),氮掺杂材料中吡啶氮能有效降低其过电势,提高ORR起始电位[30-31]. 此外,EIS图谱曲线上半圆部分直径代表电荷转移阻抗,即电子传递阻力[35].结果显示,750/N@C-CC的电荷传递阻抗在3种材料中最低,说明其电化学活性更加优异.

图5 X/N@C-CC的LSV曲线及电化学交流阻抗(EIS)谱图Fig.5 LSV curves and EIS plots of X/N@C-CC
2.3 H2O2产率
为进一步考察不同煅烧温度对材料实际电催化产H2O2能力的影响,并摸索原位产生H2O2的最佳试验条件. 以X/N@C-CC为阴极,MMO为阳极构建两电极恒流体系,在恒流情况下测定了溶液中H2O2浓度. 如图6所示,750/N@C-CC作为阴极催化剂,最高H2O2产率达73.09 mg/(L·h),为三组样品中最高;纯碳布最高H2O2产率仅为15.29 mg/(L·h),说明氮掺杂极大地增强了阴极2e-ORR性能. 通过对照试验,确定了H2O2主要来源于阴极氧还原反应. 另外,H2O2产率还受电流大小及pH的影响. 数据显示,750/N@C-CC阴极在pH为6、电流大小为20 mA时表现出最佳的2e-ORR性能. 低pH时H2O2产率较低,可能是因为析氢副反应的加剧〔见式(4)〕[36],高pH时产率较低,主要是因为H2O2属于二元弱酸,碱性条件下易电离成H2O和O2〔见式(5)〕[22]. 尽管如此,750/N@C-CC在pH=3~10的宽域内最低仍达到了47.06 mg/(L·h)的H2O2产率,证明其具有良好的电催化产H2O2性能. 需要说明的是,我们只是对反应初始pH进行了控制,由于单室中阳极的存在,体系中会同步发生析氧反应,阴阳极的协同作用导致反应体系中的pH不会发生较大变化. 控制pH为6时,当电流从10 mA增至20 mA,750/N@C-CC试验组的H2O2产率从62.68 mg/(L·h)增至73.09 mg/(L·h),但当电流继续增大时,H2O2产率出现明显降低,这是因为电流增大会导致H2O2在阴极的还原反应〔见式(6)〕,析氢副反应〔见式(4)〕及水还原反应〔见式(7)〕的出现或加剧[37]. 另外,通过与近期文献中电催化产H2O2的自支撑碳基阴极材料对比(见表2),发现750/N@C-CC在宽pH域内均具有优秀的电催化产H2O2的能力及相对高的库伦效率.
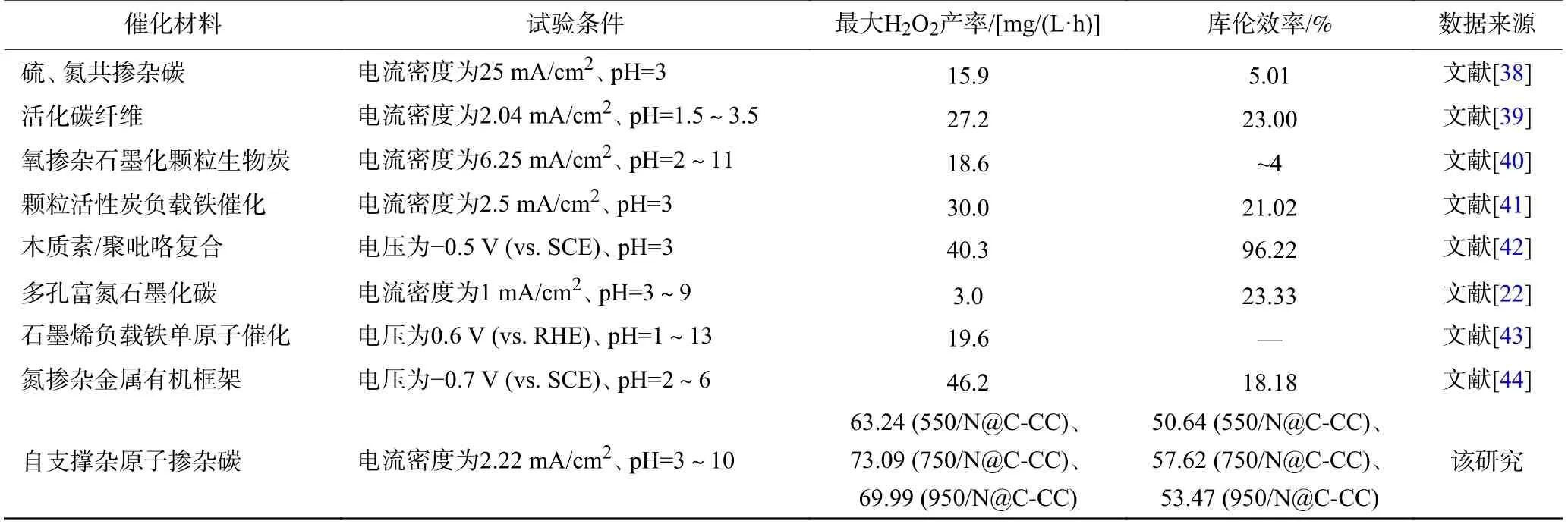
表2 自支撑碳基2e- ORR阴极材料最大H2O2产率及库伦效率的对比Table 2 Comparisons of the concentration of electrogenerated H2O2 and coulombic efficiency of the carbon-based 2e- ORR cathode materials

图6 电催化合成H2O2条件试验Fig.6 Experiments of electrocatalytic synthesis of H2O2
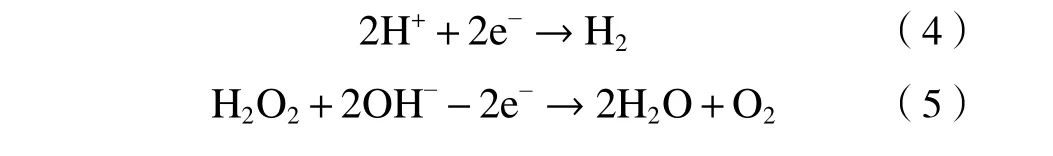

2.4 CBZ降解
在上述研究基础上,通过利用原位电催化合成H2O2,耦合UV建立高级氧化体系,考察其对CBZ的降解能力,以判定合成材料的应用可能性. 如图7所示,CBZ初始浓度为10 μmol/L,该体系在pH为3、6、10的条件下均能在20 min内将CBZ降解完全. 另外,为排除UV光源及阳极氧化的单独作用,分别进行对照试验,结果显示,pH为3、6、10时单独UV照射几乎不会使CBZ发生降解,而阳极氧化对CBZ降解的贡献最高只有33%,说明该体系下CBZ降解主要依靠UV活化H2O2产生的氧化活性物种. 此外,通过以TBA为·OH掩蔽剂来判断·OH在CBZ降解过程中的贡献,结果显示,当加入10 mmol/L TBA后CBZ降解效果几乎与阳极氧化的效果一致,说明UV耦合电催化产H2O2降解CBZ体系中主要的氧化活性物种为·OH. EPR结果显示,UV活化H2O2样品存在明显的DMPO-OH加合物的四级特征峰,进一步表明该体系下产生的氧化活性物种主要为·OH[44-46].
CBZ降解循环试验很好地证实了750/N@C-CC电催化产H2O2性能及耦合UV活化体系对CBZ降解性能的稳定性. 如图7所示,该体系在循环10次后降解能力无明显变化. 此外,在循环试验过程中,未观察到电极溶出的现象,表明该自支撑阴极材料具有较好的电化学及机械结构稳定性. 通过与近期电芬顿及UV/H2O2高级氧化体系降解CBZ的研究对比(见表3),发现UV耦合电催化产H2O2高级氧化体系具有更快的CBZ降解速率、更宽的工作pH域、无需外加H2O2等优势,使其更适用于对CBZ的降解.

图7 UV/H2O2体系下CBZ降解及自由基鉴别试验Fig.7 Degradation of CBZ and Identification of free radicals in the UV/H2O2 system

表3 CBZ在不同体系下降解情况的对比Table 3 Comparison of CBZ degradation at different systems
3 结论
a) 通过以Cd为牺牲金属,成功于碳布基底上原位合成了新型自支撑氮掺杂阴极材料. 经分析,750/N@C-CC表面拥有丰富吡啶氮、石墨氮及含氧官能团等2e-ORR活性位点. 煅烧温度不同导致掺杂氮元素的总量及类型发生较大变化,其是导致材料ORR性能变化的内因,同时含氧官能团的存在确保了催化剂材料在中碱性条件下的高H2O2产率.
b) 通过对750/N@C-CC电催化产H2O2能力进行一系列评估,结果显示,催化剂材料在pH=3~10的宽域内均具有优异的H2O2产率.
c) 构建了基于750/N@C-CC的宽pH域电催化产H2O2耦合UV的高级氧化体系,用于CBZ降解,并对其降解能力进行一系列评估. 结果表明,UV耦合电催化产H2O2体系能够有效去除CBZ,且主要活性物种为·OH. 由此,笔者认为,针对以CBZ为代表的新型有机污染物处理,以750/N@C-CC为阴极电催化产H2O2并耦合UV活化体系有着较好的应用前景.
猜你喜欢
中国煤炭地质(2022年10期)2022-11-23
油气田地面工程(2022年8期)2022-10-02
有色设备(2022年2期)2022-08-06
城市道桥与防洪(2022年3期)2022-05-08
安全与环境工程(2021年2期)2021-04-02
军民两用技术与产品(2021年10期)2021-03-16
煤炭加工与综合利用(2020年6期)2020-07-17
电子制作(2018年12期)2018-08-01
分析化学(2018年12期)2018-01-22
