
胶乳增强免疫比浊法检测抗环瓜氨酸肽抗体试剂盒的制备及性能评价
2022-07-25 15:10:26金耀建黄信用高素华楼宏强张艳芳孙义凯
金华职业技术学院学报 2022年3期
金耀建,黄信用,高素华,叶 均,楼宏强,张艳芳,孙义凯
(1.金华职业技术学院,浙江金华 321007;2.金华市强盛生物科技有限公司,浙江金华 321000)
抗环瓜氨酸肽抗体(ACPA)是在类风湿关节炎(RA)患者血清中发现的一种自身抗体,对RA 诊断具有高度特异性,检测ACPA 已成为诊断RA 的主要指标,并且除类风湿因子(RF)外,ACPA 也纳入了新版RA分类标准[1]。ACPA 在70%~80%的RA病人体内都能被检测到且特异性高达98%左右[2-3],且ACPA可以在RA患者出现临床表现前5年~10年出现,能预测更严重的临床症状[4-7]。
目前,ACPA 的检测方法有酶联免疫吸附试验(ELISA)、化学发光免疫分析法(CIA)、电化学发光免疫分析法(ECLIA)、胶体金免疫层析法(GICA)和时间分辨荧光免疫分析法(TRFIA)等,依据检测原理的差异各检测方法具有不同的优缺点。胶乳增强免疫比浊法检测ACPA是一种新型的、特异性强、灵敏度高、成本低、试剂通用型的检测方法,能早期诊断RA。为此,该检测试剂盒的制备具有十分重要的意义。
1 材料与方法
1.1 材料
羧基微球(上海辉质生物科技有限公司)、牛血清白蛋白(BSA,盐城赛宝生物科技有限公司)、环瓜氨酸肽和抗环瓜氨酸肽抗体(均为广东菲鹏生物有限公司)。
1.2 方法
该试剂盒由试剂一、试剂二、标准品和质控品四部分组成。
1.2.1 试剂一配方
磷酸氢二钠22.4 g、磷酸二氢钾5.4 g、PEG6000 20 g、氯化钠20 g、JX515 0.2 mL、纯化水1000 mL,加纯水至1068 g,密度为ρ=1.068,pH值为7.50±0.10。
1.2.2 试剂二配方
纯化水130 mL、硼酸缓冲液40 mL(硼酸1 g、纯化水40 mL,pH 值为10.00±0.10,密度为ρ=1.025)、羧基微球10 g、EDC 溶液5 mL(EDC 0.05 g、纯化水5 mL,密度为ρ=1.010)、环瓜氨酸肽2 g、BSA 溶液15 mL(BSA 1.5 g、纯化水15 mL,密度为ρ=1.100),试剂二稀释液1000 mL(磷酸氢二钠22.4 g、磷酸二氢钾5.4 g、JX515 0.2 mL,加水到1028 g,密度为ρ=1.028,pH值为7.50±0.10)。
1.2.3 反应体系中主要成分筛选
选择粒径分别为70nm、108nm及240nm的羧基微球与抗原,分别按5∶1、4∶1、3∶1、2∶1比例混合,目的是选择抗原与羧基微球交联的合适比例。羧基微球与抗原交联时间分别为30 min、45 min、60 min;BSA 封闭时间分别为30 min、60 min、120 min。将 约为100 U/mL 的ACPA 样本进行1/2 及1/4 梯 度稀释。
基本操作:样本5μL,加试剂一150 μL,混匀,37 ℃孵育300 秒,在波长700 nm 处读取吸光度A1,然后加试剂二50 μl,37 ℃孵育300 秒,在波长546 nm处读取吸光度A2,并与标准曲线比较,即能得出样本中ACPA的含量。
1.2.4 与同行试剂比对
用上述确认体系与工艺配制试剂,同时选择北京九强生物技术股份有限公司(北京九强)ACPA试剂及血清样本40 例(ACPA 含量为1 U/mL-100 U/mL)进行含量检测比对,初步确定试剂盒准确性。
1.2.5 抗环瓜氨酸肽抗体水溶液检测
校准品及质控品主要原料为ACPA,将ACPA按理论值配制成100 U/mL的水溶液,将该水溶液稀释成1/2及1/4,再用上述确认的试剂进行检测。
1.2.6 校准品溯源
依据GB/T 21415-2008/ISO 17511:2003“体外诊断医疗器械,生物样品中量的测量,校准品和控制物质赋值的计量学溯源性”要求对工作校准品和产品校准品溯源。
1.2.6 .1工作校准品溯源
选用研制的ACPA 试剂,批号为20 180903;待定值工作校准品,批号为20 181014;北京九强ACPA试剂及配套校准品、质控品,批号为20180807,校准品标示值为100.0 U/mL±11.2 U/mL,质控品靶值为30.0 U/mL±6.0 U/mL 或60.0 U/mL±12.0 U/mL;新鲜血清样本40份,在贝克曼AU480自动生化分析仪上进行检测。
建立北京九强参照系统。用北京九强配套校准品校准北京九强试剂,检测北京九强配套高低值质控品,建立参照系统(要求质控值在范围内且CV≤5.0%)。
工作校准品初定值。随机抽取10 瓶工作校准品,用以上系统检测,每瓶检测3 次,计算均匀性及均值。若均匀性符合0<F≤F0.05(vbb,vwb)的要求,则其均值作为初定值。
工作校准品终定值。在指定检测仪器上开通两个通道,一个通道为北京九强参照系统,另一个通道为初定值工作校准品,用以校准本研制试剂的定值系统,两个系统同时对40份新鲜临床样本进行检测(覆盖本研制试剂线性范围),以参照系统检测结果为x,以定值系统检测结果为y,进行线性回归y=a+bx分析,若满足以下三个条件,即斜率b在1.00±0.05 范围内,截距a在0±10 范围内;相关系数r≥0.975,则认为初定值可靠并作为该工作校准品最终定值。
工作校准品不确定度。工作校准品不确定度包括北京九强校准品引入,定值精密度引入,均匀性引入。北京九强校准品引入的相对不确定度(Uwcal)为5.60%,直接引用。定值精密度引入的相对不确定度(Urep)用北京九强检测系统测试工作校准品10次,计算公式为:
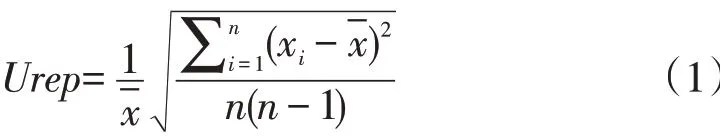
其中x为每次测定值,n为测定次数。均匀性引入的相对不确定度(Ubb)通过随机抽取10瓶工作校准品,用以上系统检测,每瓶工作校准品检测3 次,计算其均值xˉ、瓶间均方(MSbb)及瓶内均方(MSwb),从而得到Ubb:
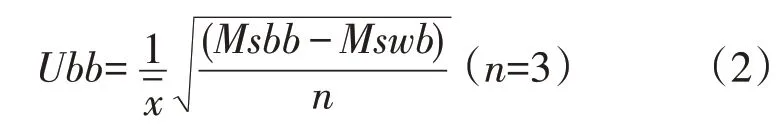
通过上述计算,进而得到总相对不确定度(Uc)及工作校准品扩展不确定度(U):
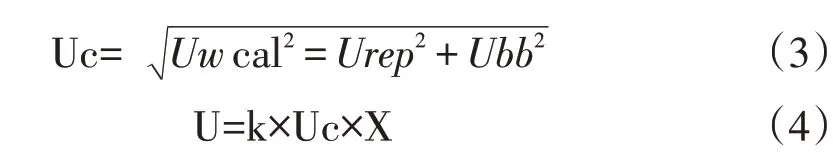
其中k=2(95%);X为工作校准品定值。
1.2.6 .2 产品校准品溯源
本研究ACPA 试剂,批号为20 181019;工作校准品,批号为20170611,定值为100.0 U/mL±11.8 U/mL;待定值产品校准品,批号为20 181019;在贝克曼AU480自动生化分析仪上进行检测。
产品校准品定值和不确定度。用工作校准品校准本试剂建立检测系统。随机抽取规格为5.0 U/mL及100.0 U/mL的产品校准品各10瓶,每瓶检测3次,计算其均匀性及均值。若均匀性符合为0<F≤F0.05(vbb,vwb)的要求,则其均值作为定值。不确定度计算方法同工作校准品。
1.2.7 质控品定值
本研制ACPA 试剂(产品校准品批号为20180806,待定值产品质控品批号为20180806,报告定值质控品1 为19.99 U/mL,质控品2 为50.02 U/mL),用产品校准品在贝克曼库尔特、日立、西门子、罗氏、迈瑞、东芝、雅培、迪瑞等品牌的21 种机型上定标,测试两水平质控品各10 次,分别计算均值作为该机型上质控品定值。最后将所有机型定值汇总,取统计学平均值作为质控品报告定值,要求质控品准确度指标与标示值偏差≤20%。
1.2.8 确定参考区间
收集宁波市开发区中心医院检验科新鲜体检血清标本240 例,其中男120 例,女120 例。排除标准:(1)肝、胆、胰、肺及心脑血管等相关指标异常标本;(2)溶血及脂浊标本;(3)放置时间超过24 小时标本。所有标本收集后,在2 ℃~8 ℃下冷藏运输。用本研制的试剂、标准品和质控品(批号:20181019)进行检测,用SPSS 22.0进行数据分析,采用Dixon 法判断离群值,采用Z 检验判断参考区间是否需分组,用Kolmogorov-Smirnov 法进行数据正态性检验,P>0.05 表示数据呈正态分布。若数据呈正态分布,采用95%可信区间计算结果X±1.96SD。
1.2.9 产品校准品和质控品稳定性
试剂稳定性主要为效期稳定性及开瓶稳定性。校准品批号为20171129,质控品批号为20171129。
1.2.9 .1 效期稳定性
校准品和质控品按照规定条件下2 ℃~8 ℃贮存,各取出2瓶放入对照温度为-80 ℃的环境中,直到12 个月稳定期满后使用同步法进行稳定性研究。使用该项目常规试剂盒,在全自动生化分析仪上重复条件下每瓶检测3 次,记录结果并用SPSS 22.0软件统计分析。
1.2.9 .2 首次开封后稳定性
校准品和质控品首次开封后按照规定条件下2 ℃~8 ℃贮存,分别在贮存0、4、8、12、16 天取出同一瓶测量,每瓶3 次,记录结果并用SPSS 22.0 软件统计分析。
1.2.10 干扰物研究
选择混合血清一份(样本批号分别为:20170905、20171017、20171129),分成五份,四份加入干扰物作为干扰标本,另一份不加干扰物作为基础标本,各干扰组标本组合见表1。每份标本重复测五遍,记录试验结果,计算分析干扰物浓度,每月测1次连续测3个月,其中各组干扰值=各组干扰物标本测定均值-基础标本测定均值。干扰值落在基础样本值±1.96SD 范围内(95%可信区间内)为无显著干扰。

表1 各干扰组标本组合
1.2.11 临床评价
收集来自于株洲市中心医院的临床血清标本100例,要求排除严重浑浊、脂血、溶血、高胆红素样本。参照试剂为浙江夸克生物科技有限公司,批号为190628。目的是验证考核试剂与已上市的对照系统等效性。对样本的检测结果进行离群值分析、回归分析、偏倚分析等指标的计算,从而评价考核试剂在临床上的安全性和有效性。
1.2.11.1 离均值分析
以浙江夸克生物科技有限公司生产的试剂和方法作为参照标准,用其与本文研制的试剂和方法,同时测定同一份血清标本,进行结果的离均值分析,以此来检验本研制的试剂和方法与参照标准的等效性。若两种方法测试的绝对差值/相对差值均超出所有样本均值的4倍,则被判断为离群值,离群值不超过总数的2.5%。
1.2.11 .2 回归分析
以考核试剂作Y轴,对照试剂作X轴,建立直线回归方程y=a+bx,计算相关系数或决定系数并计算斜率及截距的95%可信区间,相关系数要求r≥0.975。将考核试剂测定值与对照试剂测定值按照统计学方法分别进行相关性和一致性分析,以考查考核试剂与对照试剂是否等效。
1.2.11 .3 偏倚分析
计算医学决定水平的预期偏倚及95%可信区间,与可接受偏倚的限值15%进行比较,如果预期偏倚的95%可信区间未超出可接受偏倚的限值,说明二者等效。偏倚分析用方程式B=Σ(Yi-Xi)/N分别计算每组的平均偏倚B及考核试剂与对照试剂测定值差值的标准差(其中B是适当浓度范围内估计的预期(平均)偏倚,X为对照试剂测定值,Y为考核试剂测定值)。计算医学决定水平(Xc)预期偏倚估计值(B^c)及其95%区间
2 结果
2.1 反应体系中主要成分筛选结果
羧基微球粒径108 nm,微球与抗原比例3∶1,微球与抗原交联时间60分钟,BSA 封闭时间60分钟,这时方法学灵敏度最佳。结果见表2。
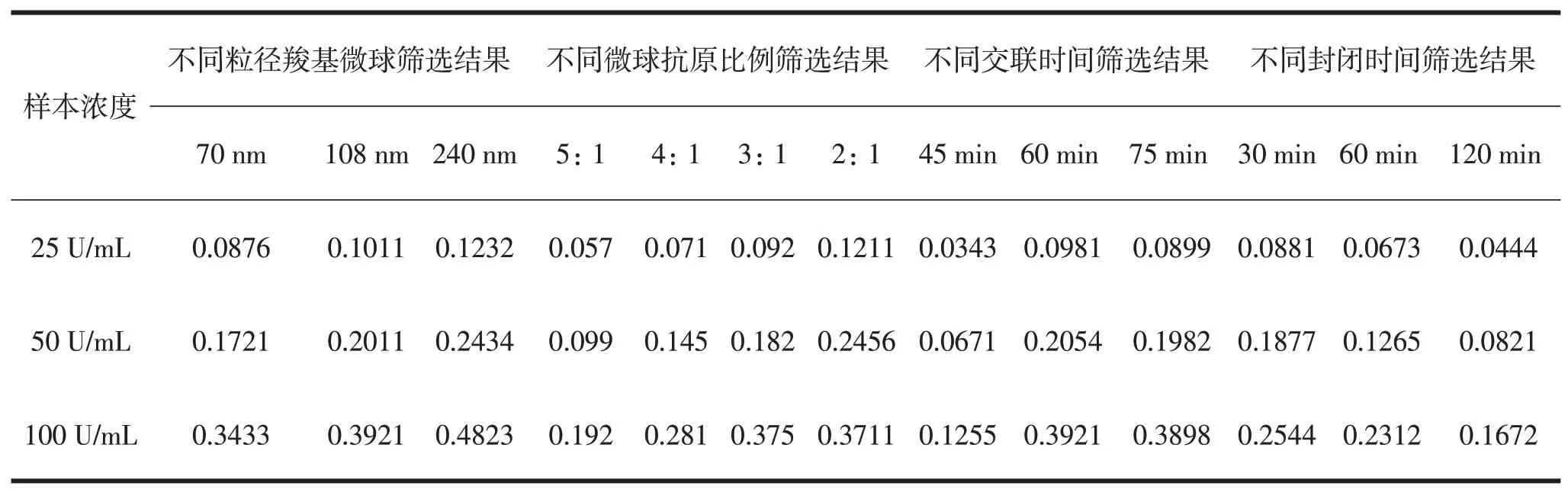
表2 反应体系中各成分筛选结果
2.2 与同行试剂比对
结果显示相关系数r=0.9835(y=0.9533x+2.5397,R2=0.9672)。因此,可以认为本试剂与北京九强试剂有很好的相关性,同时本试剂可以溯源至北京九强ACPA试剂。
2.3 抗环瓜氨酸肽抗体水溶液检测
如表3 所示,表明各浓度点实测结果与理论值偏差较小。因此,可以用抗环瓜氨酸肽抗体配制成合适浓度校准品及质控品以满足实际使用。
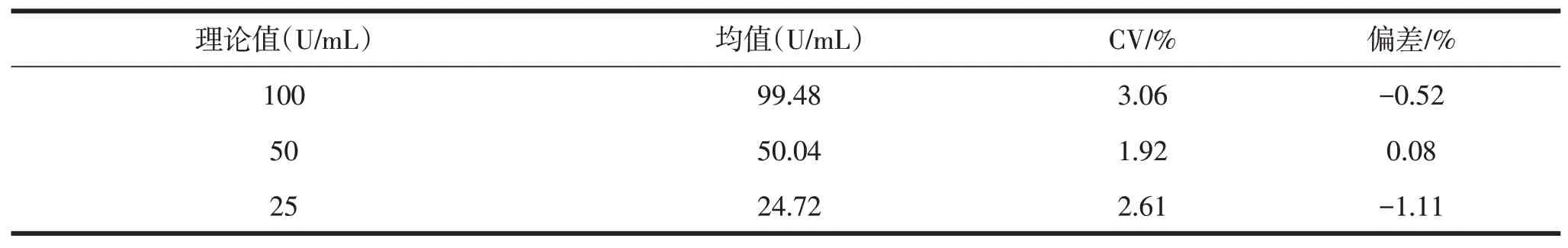
表3 抗环瓜氨酸肽抗体水溶液检测结果
2.4 校准品溯源
2.4.1 工作校准品溯源结果
以北京九强作为参照系统,溯源结果如下:北京九强低值质控品(30 U/mL)和高值质控品(60 U/mL)的均值分别是30.01 U/mL 和60.43 U/mL,偏差分别是0.04%和0.72%,CV分别是1.44%和1.10%,本工作校准品均值是100.3 U/mL,偏差0.00%,CV为1.73%,Urep=0.379%,Ubb=1.818%,Uc=5.902%,扩展不确定度=11.8。本工作校准品初定值为100.0 U/mL。参照系统和定值系统检测临床标本,线性回归统计分析结果为y= 1.010x-0.420(r=0.981),初定值符合要求。因此,本工作校准品最终定值100.0 U/mL,该批工作校准品定值为100±11.8 U/mL。
2.4.2 产品校准品溯源结果
该批产品校准品(5.0 U/mL)定值为5.0 U/mL±0.7 U/mL,产品校准品(100.0 U/mL)定值为100.0 U/mL±12.7 U/mL。结果见表4。
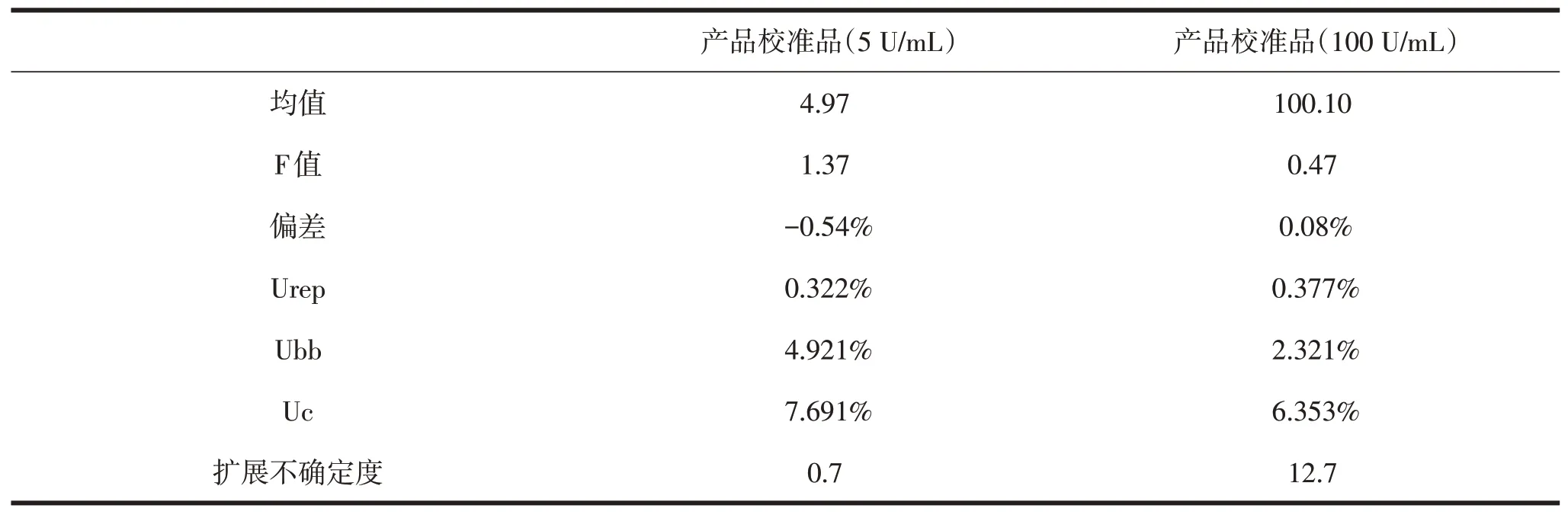
表4 产品校准品溯源结果
2.4.3 质控品定值
经过对8 个品牌的21 种机型质控品检测结果进行统计学分析,根据质控品准确度指标,该质控品定值分别为20.0 U/mL±4.0 U/mL,50.0 U/mL±10.0 U/mL。
2.4.4 参考区间
对240 名临床血清标本数据分析(见表5)结果表明:(1)Z<Z*,则无需进行男女分组;(2)合并男女分组后进行正态性检验,结果显示符合正态分布;(3)合并男女分组后计算参考上限及下限,则参考下限及上限分别为5.0 U/mL及34.3 U/mL。因此,参考区间确定为0 U/mL~35 U/mL。

表5 血清检测结果统计学分析
2.4.5 产品校准品和质控品稳定性
产品校准品及质控品在2 ℃~8 ℃贮存0个月、3 个月、6 个月、9 个月、12 个月,效期稳定性检测结果变化趋势不显著(P≥0.05)。
产品校准品及质控品首次开封后稳定性检测,0 天、4 天、8 天、12 天、16 天检测结果的变化趋势不显著(P≥0.05)。
2.4.6 干扰物研究
胆红素干扰试验、甘油三酯干扰试验、血红蛋白干扰试验结果经统计学分析,当胆红素≤200 mg/L、甘油三酯≤3 g/L、血红蛋白≤2.5 g/L 时,对三批试剂结果均无显著影响。
2.4.7 临床评价
用对照试剂和考核试剂检测临床血清标本100例,离群点数量为0 例;两者回归分析结果y=0.9607x-0.3451(P<0.05),系数b 95%可信区间CI=0.9180~1.0034(P<0.05),截距a 95%可信区间CI=-2.4020~1.7118(P≥0.05),说明两种试剂直线关系成立。
对考核试剂和对照试剂检测结果进行一致性分析,两者检测结果的Bland-Altam 分析基本数据见表6,绝对偏倚和相对偏倚都在允许范围内。
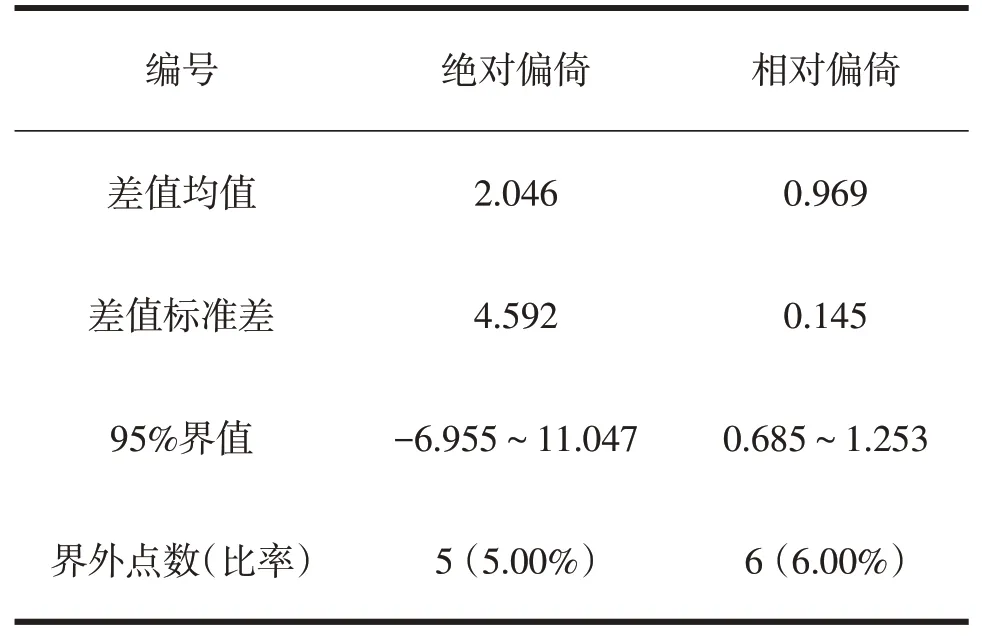
表6 Bland-Altam分析基本数据
偏倚分析结果,当Xc=35 U/mL 时,B^c95%区间为[-2.639,-0.8022],而允许限度为[-5.25,5.25],考核试剂相对于对照试剂检测结果,偏倚量在允许误差范围内。
3 讨论
胶乳增强免疫比浊法是近年来快速发展的一种稳定、结果准确、能自动化、微量化、快速化检测体液蛋白均相免疫比浊的方法[8-10],该方法是在羧基微球表面交联环瓜氨酸肽抗原成为致敏颗粒,增加了灵敏度。检测患者血清中ACPA 时,当抗原致敏的微球和病人血清中的ACPA 结合并聚集在一起,改变了反应体系中液体的透光性,透光性(即吸光度)的变化与反应体系中的抗体含量呈线性相关,通过标准曲线即可得出患者血清中的抗体含量。
通过对本研制的试剂盒反应体系中主要成分筛选,确定了最佳方法学,并将试剂盒与北京九强ACPA 试剂盒比较,结果为y=0.9533x+2.5397,R2=0.9672,说明两者具有很好的相关性。
溯源性是试剂盒能否用于临床诊断的重要环节,本文以北京九强作为参照系统,将参照系统与本工作校准品定值系统检测临床标本结果进行统计分析,无论均值偏差还是变异度都符合要求,线性回归y=1.010x-0.420(r=0.981)。
该方法灵敏、稳定,适用于临床常用机型,最低检测下限为5.0 U/mL,线性范围较宽,参考范围95%可信区间为<35 U/mL。产品校准品和质控品稳定性好,在2 ℃~8 ℃贮存12 个月和开封16 天后检测结果变化不显著。干扰物胆红素≤200 mg/L、甘油三酯≤3 g/L、血红蛋白≤2.5 g/L 时,对三批试剂结果均无显著影响。在临床评价中,对照试剂和考核试剂检测100例临床标本,线性回归y=0.9607x-0.3451(r=0.9763),偏倚分析在允许误差范围内,说明考核试剂完全达到临床检测要求。本研制的试剂盒已取得中华人民共和国医疗器械注册证(体外诊断试剂),注册证编号:浙械注准20202400782。
猜你喜欢
中学生数理化(高中版.高二数学)(2022年1期)2022-04-26 13:59:54
新世纪智能(教师)(2021年2期)2021-11-05 08:43:26
潍坊学院学报(2020年6期)2020-11-22 08:04:10
电子制作(2018年10期)2018-08-04 03:25:02
电子制作(2018年12期)2018-08-01 00:48:08
现代检验医学杂志(2016年1期)2016-11-12 13:19:34
中国免疫学杂志(2016年2期)2016-01-30 21:18:21
大连工业大学学报(2015年4期)2015-12-11 04:06:50
中国当代医药(2015年29期)2015-03-01 02:07:41
疑难病杂志(2014年12期)2014-04-16 05:19:33
