
液质联用测试尿样中的氟胺酮
2022-07-25 07:46张婷林必成
当代化工研究 2022年13期
*张婷 林必成
(福建正中司法鉴定所 福建 350003)
1.氟胺酮的来源及性状
国内最早于1987年[4]报道了氟胺酮合成方法,其目的是作为麻醉药物研究,氟胺酮的化学合成过程与氯胺酮相似。国外在2014年也有氟胺酮作为氯胺酮的衍生物进行研究合成的报道[5]。
氟胺酮的分子式为C13H10FNO,分子量为221.2,化学名:2-(2-氟苯基)-2-甲基氨基-环己酮[3],其化学结构式如图1,其外观为白色结晶粉末,与氯胺酮相似。氟氨酮作为氯胺酮的替代物或掺入氯胺酮之中被犯罪分子进行非法吸食,在(氟氨酮被列管前)以期逃避法律制裁。

图1 氟胺酮结构式
2.实验部分
(1)仪器
岛津高效液相色谱LC-20AD与三重四级杆质谱仪LCMS-8045联用系统。
(2)试剂
①氟胺酮标准溶液(1mg/mL);
②氟胺酮标准储备液:将1mg/mL氟胺酮标准溶液用甲醇稀释至0.1mg/mL作为标准储备液,实验中所用其他浓度标准工作液均由此标准储备液用纯水稀释得到;
③乙腈(默克,HPLC);
④甲醇(默克,HPLC);
⑤甲酸(阿拉丁,HPLC);
⑥纯水(屈臣氏)。
(3)氟胺酮液质联用方法优化
①用LCMS-8045做Q3扫描(图2氟胺酮一级质谱图),一级质谱检测质量范围m/z100~300,确定氟胺酮母离子为222.1。

图2 氟胺酮一级质谱图
②使用设备软件自动优化产物离子及MRM参数(图3氟胺酮产物离子扫描质谱图),确定氟胺酮产物离子及电压(表1氟胺酮MRM参数)。
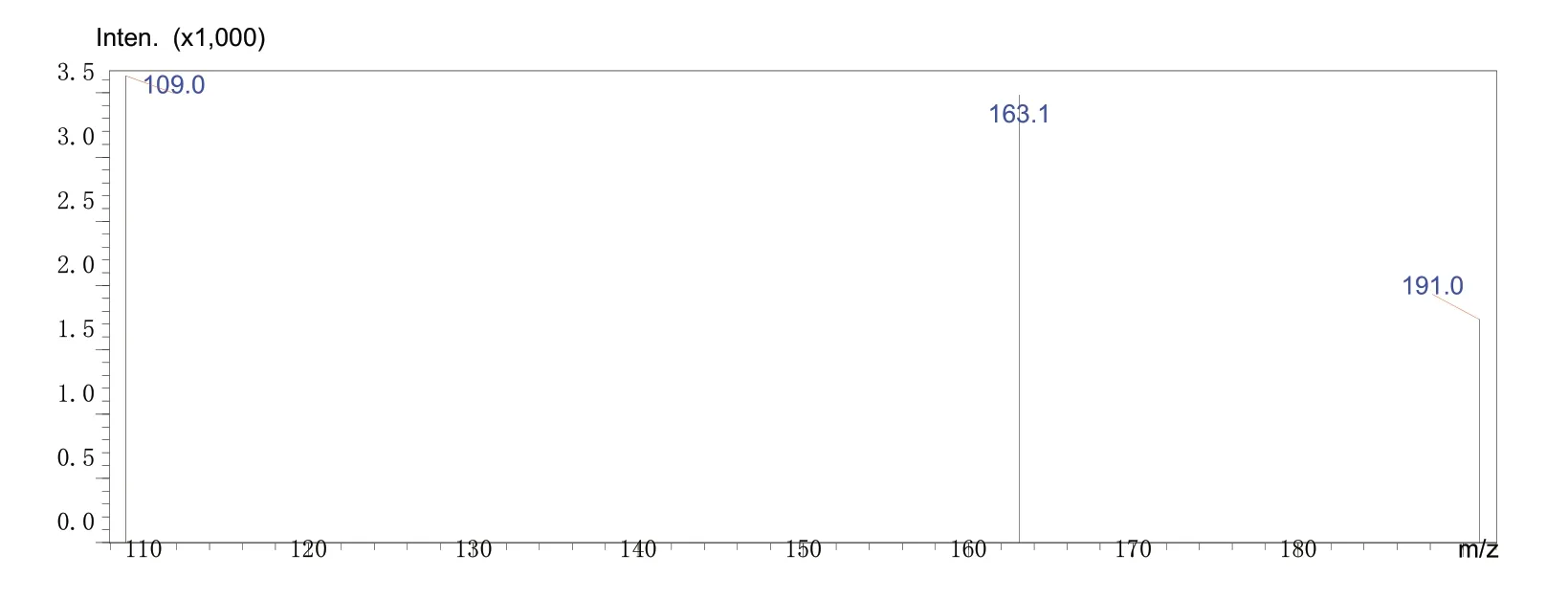
图3 氟胺酮产物离子扫描质谱图
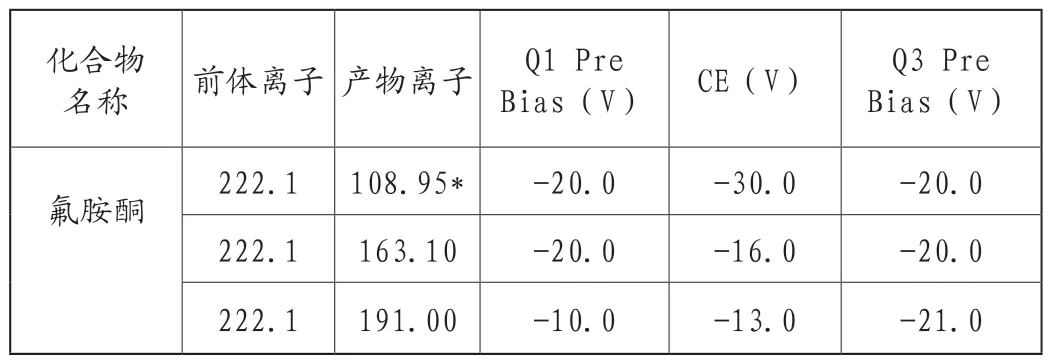
表1 氟胺酮MRM参数
(4)样品制备
①标准溶液配制
将0.1mg/mL的氟胺酮标准储备液,用水逐级稀释成浓度为0.1μg/mL、0.2μg/mL、0.5μg/mL、1μg/mL、2μg/mL、5μg/mL、10μg/mL的标准工作液用于建立标准曲线。
②样品前处理
采集正常人尿样加标样分别配制成浓度为1ng/mL、2ng/mL、5ng/mL、10ng/mL、20ng/mL、50ng/mL、100ng/mL的加标样后离心,取上清液过0.22μm微孔滤膜后直接用于液质分析。空白基质采用尿样不加标的方式,其余处理步骤相同。
(5)实验室测试条件
①LC(液相)条件
液相(LC):LC-20AD;
液相分析柱:岛津Shimadzu Shim-pack GIST C18(1.9μm×2.1mm×100mm);
流动相:A-0.1%甲酸:水,B-乙腈;
洗脱:梯度洗脱(梯度洗脱条件见表2);初始流动相:80%流动相A,20%流动相B;

表2 氟胺酮梯度洗脱条件
流速:0.3mL/min;
进样体积:2μL;
柱温:40℃。
②MS(质谱)条件
质谱仪(MS):LCMS-8045;
分析源:ESI(+)离子源;
接口电压(离子源):4.5kV;
离子源雾化气:氮气3.0L/min;
离子源干燥气:氮气15L/min;
离子源碰撞气:氩气;
DL管(脱溶剂)温度:250℃;
加热器温度:400℃;
质谱扫描方式:MRM(多反应监测)。
3.结果与讨论
(1)标准样品的MRM色谱图
10ng/mL氟胺酮加标样MRM色谱图如图4所示。
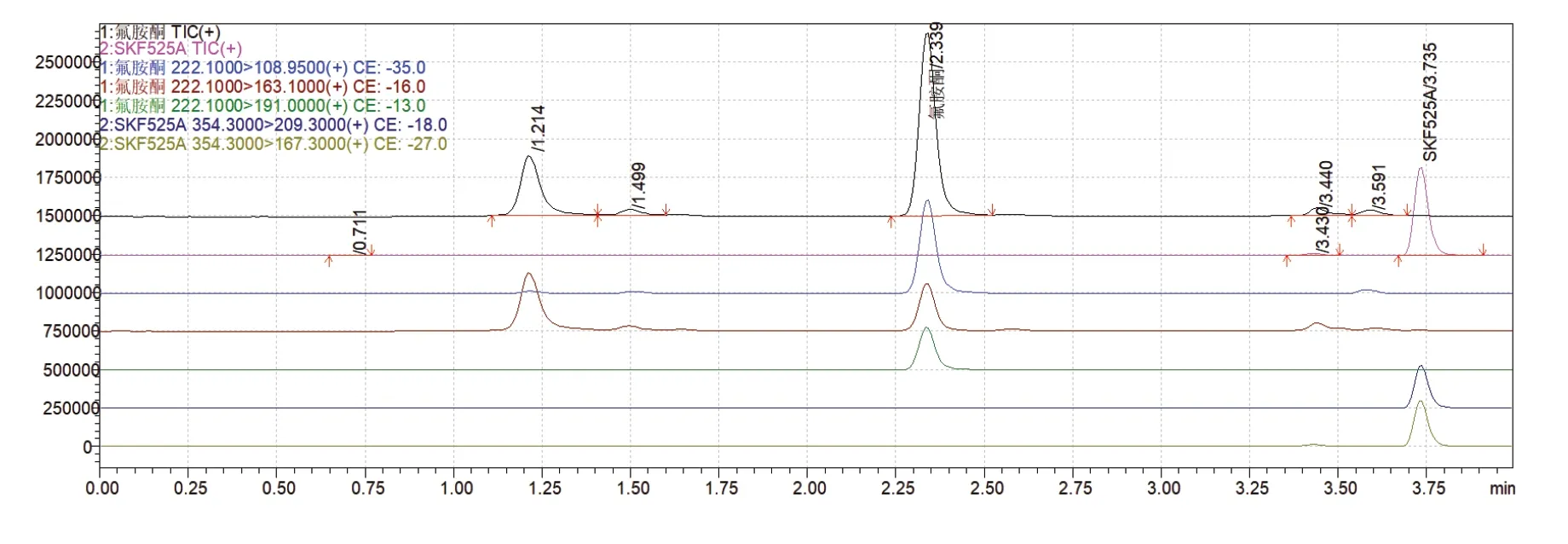
图4 10ng/mL氟胺酮加标样MRM色谱图
(2)线性关系
将浓度为1ng/mL、2ng/mL、5ng/mL、10ng/mL、20ng/mL、50ng/mL、100ng/mL的加标样按2.5的测定条件进行检测,以尿液汇总的不同浓度含量氟胺酮为横坐标,测得的氟胺酮峰面积为纵坐标,外标法(7点)建立校准曲线,如图5所示。尿样中的氟胺酮在1~100ng/mL的浓度范围内,线性关系良好,相关系数见表3。

表3 氟胺酮标准曲线
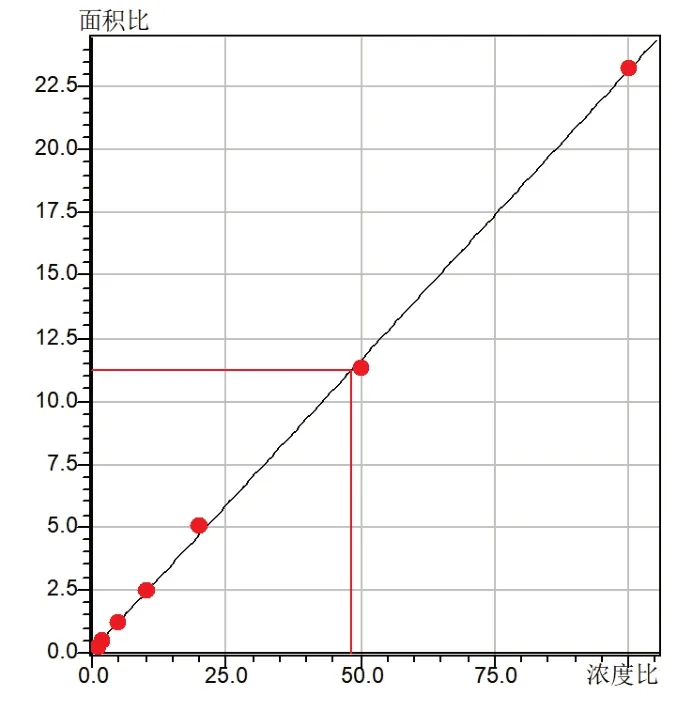
图5 氟胺酮标准工作曲线
(3)检出限和定量限
加标浓度为1ng/mL的尿样平行测定6份,仪器对氟胺酮尿样基质的方法检出限为0.05ng/mL,定量限为0.195ng/mL,表明该方法具有良好的灵敏度。氟胺酮的检出限和定量限见表4。

表4 氟胺酮检出限和定量限
(4)精密度实验
浓度为5ng/mL、20ng/mL、100ng/mL的加标样依次进样,平行测定6次,氟胺酮在不同浓度下的保留时间的相对标准偏差在0.25%~0.77%之间,峰面积的相对标准偏差分别为0.31%~3.38%之间,本液质联用仪的精密度良好(见表5)。
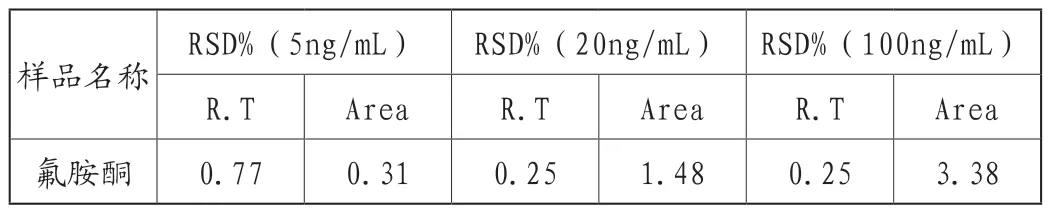
表5 氟胺酮保留时间和峰面积的重复性结果(n=6)
4.结论
本文主要研究了尿样中氟胺酮的液质联用仪检测的前处理方法及检测条件;在确定的检测条件下对方法的检出限、定量限、精密度及线性进行了考察。仪器对氟胺酮尿样基质的方法检出限为0.05ng/mL,定量限为0.195ng/mL,表明该方法具有良好的灵敏度;尿样中的氟胺酮在1~100ng/mL的浓度范围内,线性关系良好,相关系数为0.9993;氟胺酮在不同浓度下的保留时间和峰面积的相对标准偏差分别在0.31%~ 3.38%和0.25%~0.77%之间,仪器具有良好的精密度。
猜你喜欢
山东医药(2022年19期)2023-01-06
核技术(2022年7期)2022-07-22
家庭科学·新健康(2022年2期)2022-03-07
科学技术创新(2020年12期)2020-06-22
中国神经精神疾病杂志(2020年9期)2020-01-14
科技视界(2019年18期)2019-08-07
中国新技术新产品(2019年23期)2019-01-20
分析化学(2018年7期)2018-09-17
环球时报(2016-05-18)2016-05-18
分析化学(2015年4期)2015-06-08
