
引种仔猪进境及传代后肠道菌群演替发育规律
2022-07-23 07:50陈椿桦许智强刘蔓莉宋战昀吉林大学动物医学学院吉林长春3006长春海关技术中心吉林长春3006
中国兽医学报 2022年5期
陈椿桦,李 响,王 颖,许智强,李 可,刘蔓莉,王 卓,宋战昀,冯 新* (.吉林大学 动物医学学院,吉林 长春 3006;.长春海关技术中心,吉林 长春 3006)
我国既是养猪大国,也是猪肉消费大国,生猪饲养量和猪肉消费量均占世界总量的1/2左右[1]。受高生长速率、高瘦肉率等指标为主的需求导向影响,我国生猪产业的“芯片”——种质资源,约有九成以上从国外引种扩繁,2008至2017年间我国从国外共引入种猪90 384头[2],2020年数量达22 091头,创历史新高。但我国生猪市场重引进、轻选育,易陷入“引种—退化—引种”的不良循环,正遭遇“卡脖子”风险。
动物肠道微生物群的结构和组成由许多因素决定,如品种、遗传、年龄、饮食、系统发育及饲养环境[3]。不同品系肠道菌群组成不同,有研究表明中国金华猪肠道群落基本组成为厚壁菌门占70.4%、拟杆菌门占14.4%,而杜洛克猪、约克夏猪以及长白猪等西方品种猪肠道群落中厚壁菌门分别为39.6%、42.0%及45.6%,拟杆菌门分别为57.0%、51.4%及47.6%[4]。对不同遗传背景的长白猪(生长速度快的瘦肉型猪)和梅花猪(生长速度慢的肥胖型猪)的结肠中微生物组和代谢组比较分析,发现长白猪结肠中短链脂肪酸(SCFA)和次级胆汁酸含量较高[5]。
肠道微生物与预防传染病、维持肠道形态、营养消化、新陈代谢和宿主免疫调节相关联[3,6],在仔猪断奶前通过口服的方式将健康本土猪的粪便微生物群转移到商业杂交仔猪中,可降低仔猪腹泻发生率[7]。丁酸梭菌和粪肠球菌可以促进断奶仔猪的生长性能,保护肠道绒毛形态,提高免疫力,优化肠道菌群[8],许多共生细菌还可调节肠道病原体的免疫防御以及伴随它们的病理性炎症[9]。而有害的微生物群落可以改变肠道菌群的组成并引起疾病,诱发炎症性疾病,如炎症性肠病(IBD)[10]。消化道是非常复杂的微生态系统,宿主的微生物群与外部环境之间存在动态的共生关系[11]。仔猪肠道菌群的稳态受各种因素的影响,包括微生物菌群的定植和演替、猪的日龄、遗传因素、饲养环境、抗菌剂、膳食成分、饲料添加剂、饲养管理、疾病状况、断奶和季节等[12]。
我国主要引种断奶仔猪,经过纯代培养后再杂交培养,最终成为商品猪。LU等[13]在杂交猪群中收集3个时间的粪便样本,评估了每只动物粪便微生物组的组成随时间的变化,在每个时间点确定了2种肠型,发现肠型与背部脂肪厚度相关。在另一项研究中,DE RODAS等[14]研究了猪肠道微生物组在7个时间点沿不同解剖部位的纵向变化,结果显示年龄效应在采样的所有解剖部位中都很明显。WANG等[15]对猪肠道微生物群从出生到市场进行了全面的动态的纵向研究,发现不同阶段猪肠道微生物群可能由不同生长阶段的饮食和/或肠道生理学差异决定。上述研究通过年龄、饮食、解剖部位等条件的变化调查了生猪肠道微生物群的组成和变化,评估的对象涉及杂交猪和商品猪。但跨境引种生猪纯代培养期间在环境变化以及遗传繁育过程中肠道菌群的变化和维持仍不清楚。
因此,本研究旨在揭示进境仔猪在遗传及环境因素作用下肠道菌群的演替发育规律和变化趋势,找出不同品种、不同日龄肠道菌群的关键差异菌属,探讨饲养方式和环境因素对肠道菌群的影响。
1 材料与方法
1.1 样品采集及分组采集相同饲养环境、饲养方式,同一批次法国引进种猪及其后代粪便样本立即冷链运至实验室,分组标记后-80℃保存,开展猪粪便细菌16S rRNA基因的V3~V4区高通量测序,共计采集了36份合格样品,样品信息如表1所示。
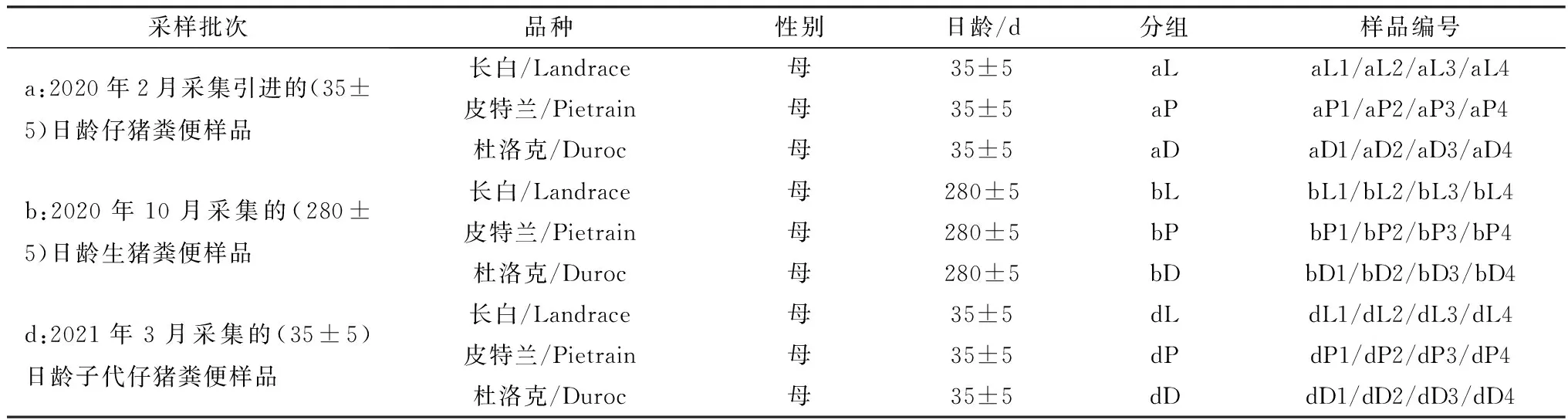
表1 样品信息表
1.2 仪器与试剂PCR扩增仪(ABI);酶标仪(BioTek);电泳仪(北京六一生物科技有限公司);凝胶成像系统(北京百晶生物技术有限公司);QIAamp粪便DNA提取试剂盒(QIAGEN公司);Q5®High-Fidelity DNA Polymerase(NEB公司);Quant-iT PicoGreen dsDNA Assay Kit(Invitrogen公司);Agarose(Invitrogen公司);Marker(TaKaRa公司);TAE(Invitrogen公司);凝胶回收纯化试剂盒(AXYGEN公司);NanoDrop(Thermo Scientific公司);TruSeq Nano DNA LT Library Prep Kit(Illumina公司);MiSeq试剂盒V3(Illumina公司)。
1.3 样品DNA提取和测序用QIAamp粪便DNA提取试剂盒提取样品微生物基因组DNA,然后用NanoDrop ND 2000分光光度计和琼脂糖凝胶电泳检测样品DNA的完整性。用Quant-iT PicoGreen dsDNA分析试剂盒和酶标仪测定基因组DNA的质量浓度,并将其稀释至20 mg/L,-20℃保存备用。
针对16S rRNA V3~V4区域设计引物,用于样品中细菌16S rRNA基因的V3~V4可变区测序,上游引物:5′-ACTCCTACGGGAGGCAGCA-3′;下游引物:5′-GGACTACHVGGGTWTCTAAT-3′。扩增体系:5 μL反应缓冲液(5×)、5 μL GC缓冲液(5×)、2 μL dNTP(2.5 mmol/L)、1 μL上游引物(10 μmol/L)、1 μL下游引物(10 μmol/L)、0.25 μL Q5®High-Fidelity聚合酶(NEB,USA)、2 μL DNA模板和8.75 μL ddH2O,共计25 μL。扩增条件:98℃预变性2 min;98℃变性15 s、55℃退火30 s、72℃延伸30 s、25个循环;72℃延伸5 min,10℃保温。用1.2%琼脂糖凝胶电泳检测PCR扩增产物,通过凝胶回收纯化试剂盒回收纯化目的片段。
1.4 测序文库建立用TruSeq Nano DNA LT Library Prep Kit(Illumina公司)制备测序文库。通过定量PCR对文库进行定量,合格的文库浓度应在2 nmol/L以上。定量后,用Illumina MiSeq平台和相应的MiSeq试剂盒V3(Illumina公司)进行2×300 bp配对末端测序。对36个DNA样品构建测序文库。
1.5 数据处理与分析
1.5.1Illumina平台对群落DNA片段进行双端测序 通过质量初筛的原始序列按照Index和Barcode信息,进行文库和样本划分,去除Barcode序列。然后按照QIIME2 dada2或Vsearch软件的分析流程进行序列去噪或OTU聚类分析。
1.5.2各组不同物种分类学水平的展示分析 使用QIIME2[16](2019.4版本)计算α多样性并绘制稀疏曲线反映测序深度是否合适。使用非量度多维尺度分析(NMDS)和非加权配对平均法(UPGMA)衡量不同组间的β多样性差异。通过R软件绘制热图进行聚类分析。在QIIME2软件中调用“qiime taxa barplot”命令,在物种分类学组成层面上进一步衡量不同组间的物种丰度组成差异,寻找标志物种。通过PICRUSt微生物代谢功能预测工具,将样本16S rRNA基因测序数据与MetaCyc (https://metacyc.org),KEGG (http://www.genome.jp/kegg/pathway.html)数据库进行比较,预测样本的菌群代谢功能,分析差异通路。
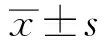
2 结果
2.1 物种分类学注释结果每个分类级别上的OTU数量统计结果如图1所示,长白猪和杜洛克猪在各分类学水平上的OUT数无差异,皮特兰猪在科水平上入境后(b组和d组)与入境时(a组)具有极显著性差异,表明皮特兰猪肠道菌群易受环境的影响。
A.物种分类学注释;B,C,D.分别为长白猪、皮特兰猪、杜洛克猪3个生长阶段各分类水平的统计学分析。图中(A)横坐标代表各组内样本(n=4)平均值,纵坐标代表注释结果中分类水平分别为门、纲、目、科、属、种的ASV/OTU数图1 物种分类学注释
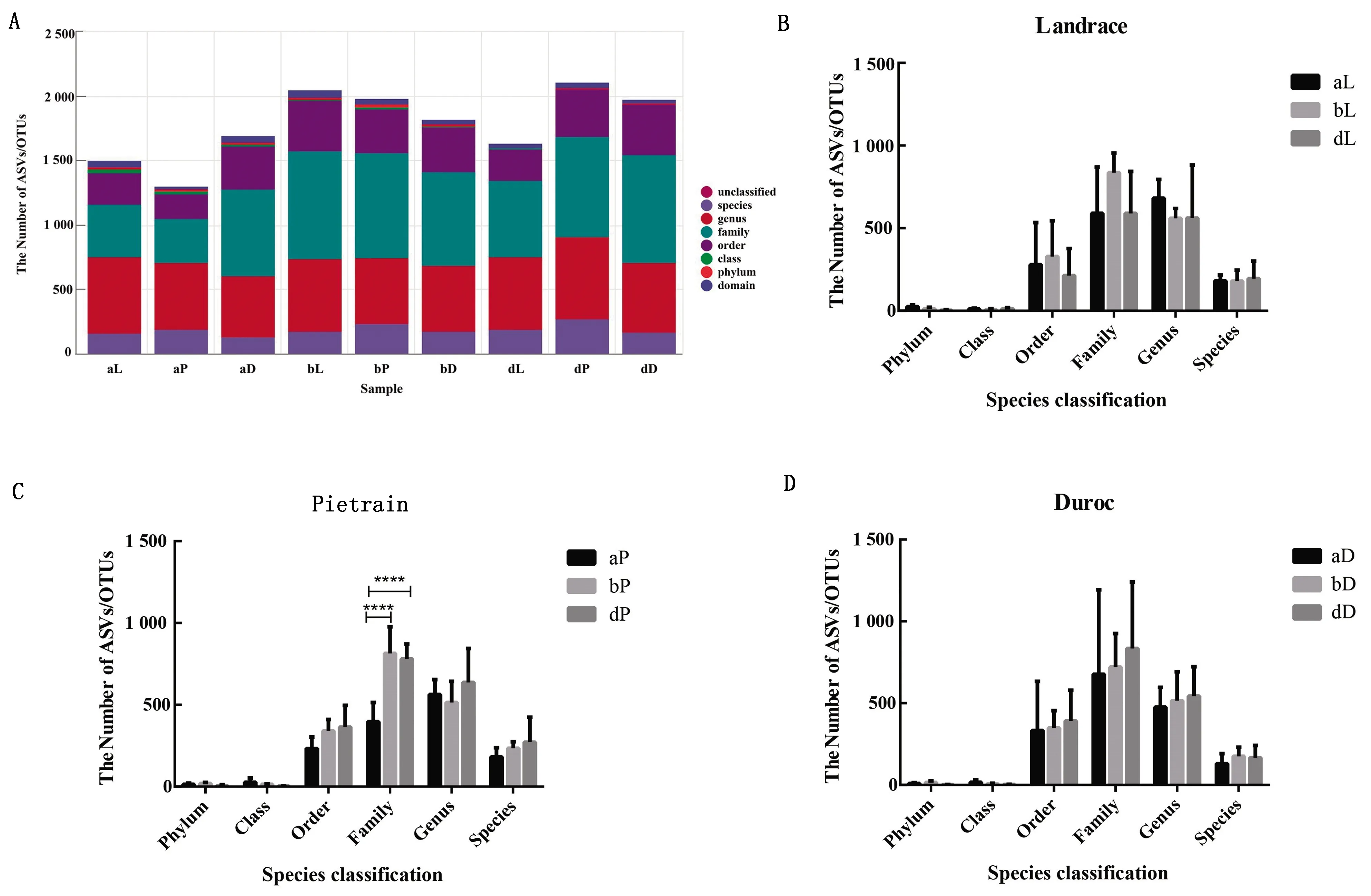
2.2 肠道微生物丰富度及多样性的变化肠道微生物丰富度及多样性图2A所示,随着样品数量的增加,稀疏曲线逐渐饱和,说明36个样品的微生物种群足以评估细菌丰富度和微生物群落多样性。α多样性指数结果如图2(B、C、D)显示,长白猪和杜洛克猪3组之间的丰富度和多样性无显著性差异。皮特兰品种猪群落的丰富度和多样性受环境影响大,入境后(b和d 2组)丰富度较入境时(a组)显著增加;在我国饲养280 d后多样性降低,但经过传代后菌群多样性增加。初步说明环境和遗传等因素对皮特兰品种猪肠道菌群丰度和多样性有显著影响。
A.稀疏曲线;B,C,D.分别为长白猪、杜洛克猪、皮特兰猪α多样性指数丰度图。图中(A)横坐标为抽平深度,纵坐标为10次计算的α多样性指数的中位值与箱线图。图中(B、C、D)每个顶端灰色区域标识对应一种α多样性指数,横坐标为分组标签,纵坐标为相应α多样性指数的值图2 稀疏曲线及α多样性分析
2.3 肠道微生物β多样性分析结果
2.3.1NMDS分析结果 NMDS分析结果如图3所示,3个品种3组样本之间的距离较大,表明入境后(b、d)与入境时(a)菌群组成差异显著。3个品种均是b组的组内样本离散度小,说明在引进仔猪后饲养一段时间,样本间菌群组成趋于相似,但在传代后(d组)样本间离散度再次变大,且长白猪的细菌群落组成最为多样化。

NMDS分析认为2点之间的距离越近(远),2个样本中微生物群落的差异越小(大),NMDS结果的应力值(Stress)小于0.2时,NMDS分析的结果较可靠图3 基于Bray-Curtis距离的NMDS分析
2.3.2层次聚类分析 UPGMA聚类分析(图4A)结果显示进境前后生猪肠道菌群的组成情况,进境前(a组)占比较高的菌属是乳杆菌属和不动杆菌属;进境后(b组)占比较高的菌属是链球菌属;传代后(d组)普氏菌、颤螺旋菌、布劳特氏菌和普拉梭菌占比接近。热图分析(图4B)结果显示前50个菌属丰度随环境的变化情况,菌群之间此消彼长,乳酸菌和不动杆菌丰度大量降低,普拉梭菌、普氏菌、颤螺菌属、布劳特氏菌属以及瘤胃球菌属大量增加,且3个品种变化趋势相同。
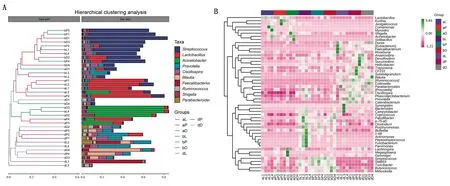
A.基于Bray-Curtis距离的层次聚类分析;B.热图分析。层次聚类分析左边的面板是样本根据彼此之间的相似度聚类,样本间的分支长度越短,两样本越相似;右图的面板为丰度排名前10的菌属堆叠柱状图。热图分析横坐标为各组样本名,纵坐标为丰度前50的菌属;右侧丰度数值条,绿色到粉色表示菌属丰度值由高到低图4 层次聚类分析及热图分析
2.4 组间菌群组成差异分析由于3个品种菌群结构的变化趋势相同,因此在组间菌群组成差异分析中不对品种进行差异分析,只对3个批次的样本进行差异分析。
在门级,OTU可以分为10个门,共享最多的OTU属于厚壁菌门、变形菌门、拟杆菌门和梭杆菌门,其中厚壁菌门、变形菌门、拟杆菌门和约占总数的90%(图5A)。统计分析图(图5B)结果显示,在仔猪进境后,拟杆菌门和变形菌门的丰度有显著差异(P<0.05)。其比例发生了变化:入境前(a组)变形菌门与拟杆菌们的比例接近于3∶1;入境后(b、d)变形菌门与拟杆菌门的比例接近1∶3,呈现完全相反的比例。且b、d 2组的变化趋势相同,即进境仔猪在我国饲养280 d后肠道菌群组成发生巨大变化,且这种变化在传代后也没有逆转。
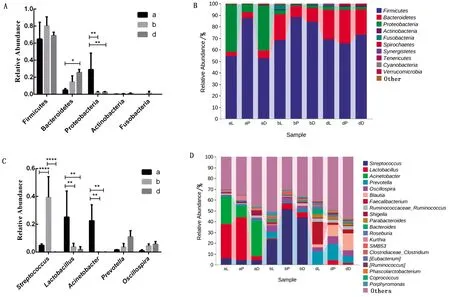
A.门水平物种丰度图;B.门水平物种差异分析图;C.属水平物种丰度图;D.门水平物种差异分析图图5 物种组成分析
在属级,如图5C所示有20个属被注释。对柱状图中丰度较高的6个菌属进行统计分析,结果如图5D所示,入境前(a组)乳杆菌属和不动杆菌属丰度最高且与其他组有显著性差异,入境后(b组)链球菌属丰度最高且与其他组有极显著差异,传代后(d组)丰度较高的菌属有普氏菌属、颤螺旋菌属、布劳特氏菌属和普拉梭菌属,但与其他组无显著差异。柱状图和差异图分析结果明显表明,在境内外饲养管理及环境条件变化下仔猪肠道菌群结构差别很大,进一步说明饲养管理及环境变化导致菌群结构的改变是导致性状退化的重要原因之一。
2.5 代谢功能预测代谢功能单元Pcoa分析结果如图6A所示,进境仔猪在我国经过1次传代后(d),代谢功能组成上与入境前(a)已几乎没有交叉。将每组与代谢相关的粪便细菌样本的注释信息与MetaCyc数据库以及KEGG数据库进行比较并进行a和d 2组的差异分析,结果显示引种仔猪传代后(d组)的视黄醇代谢极显著下调,细胞色素P450对异生素的代谢极显著下调,氟苯甲酸甲酯降解功能显著下调以及肽聚糖合成显著下调(图6B)。
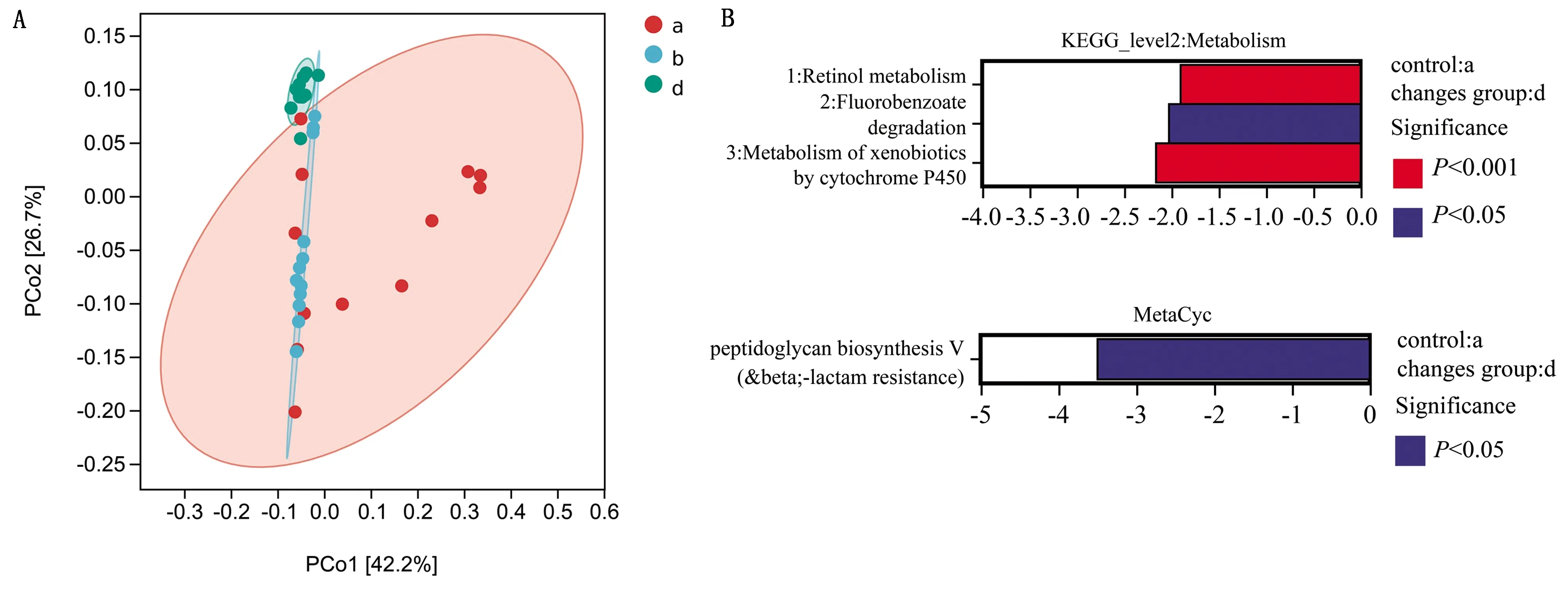
A.功能单元Pcoa分析;B.代谢通路差异分析。注:图A坐标轴括号中的百分比代表对应的坐标轴所能解释的样本差异数据的比例;2点在坐标轴上的投影距离越近,表明这2个样本在相应维度中的功能组成越相似;图B中横轴Log2(fold change)的正值代表变化组相对于对照组上调,负值为下调;纵坐标为不同的代谢通路标签图6 代谢功能预测
3 讨论
猪的肠道微生物群是一个非常复杂的生态系统,随着时间和胃肠道变化呈现出不同的动态组成和多样性[17]。仔猪在出生过程和出生后会随机接触并获取母猪产道、皮肤、粪便、母乳以及环境中的微生物,并迅速在自身肠道内定植[18],出生到断奶后2周是仔猪肠道菌群定植与初步成熟的关键时期,早期肠道菌的定植和固化会影响猪后期的生长和生产性能[17,19]。研究表明仔猪断奶后与母体分离、运输、饮食和环境条件的变化,会对仔猪产生深刻影响[20]。在跨境引种这种环境条件巨变的情况下,必然会对断奶仔猪肠道菌群产生影响,但影响的程度如何,且仔猪的变化会不会延续到下一代,还需后续深入研究。
经过对3个时间段仔猪肠道菌群的监测,我们发现,仔猪进境后经过280 d饲养,皮特兰猪肠道菌群多样性降低,但在传代后多样性再次升高,显示出环境和遗传双因素对肠道菌群的影响。进一步分析显示,在进境时至传代1次后,猪肠道菌群组成与刚进境时已完全不同,且b组和d组的变化趋势相同,d组与a组即父代与子代有显著性差异,说明环境因素对肠道菌群的影响更大,传代并没有逆转环境导致的变化。XIAO等[21]对来自法国、丹麦和中国的287头猪的粪便宏基因组进行了测序,发现性别、年龄和宿主遗传因素可能会影响猪肠道微生物组。ROTHSCHILD等[22]对1 046名拥有相对共同的环境,具有几个不同祖先起源的健康个体的基因型和微生物组数据进行分析,其研究结果表明基因型无关的个体具有相似的肠道微生物群组成,肠道微生物组与遗传祖先没有显著的关联。GOODRICH等[23]对英国的2 252对双胞胎的研究结果显示,只有1.9%~8.1%的微生物组群是可遗传的。可见宿主遗传在决定微生物组组成方面的作用很小。因此,在本研究除了环境和遗传,还考察了品种和饲料对肠道菌群的影响。BIAN等[24]使用交叉饲养模型,首次揭示了品种对新生仔猪肠道菌群发育的影响,品种影响可能在整个时期持续存在,但所有仔猪同时摄入个体母猪奶和固体饲料的复杂性稀释了品种对细菌的影响。这点在本研究热图分析中可以看出,3个品种菌群结构变化趋势相同,品种因素影响很小。
导致差异的菌属,在门水平上厚壁菌门、拟杆菌门、变形菌门3个门占比达90%以上。拟杆菌门和厚壁菌门细菌是正常寄居在哺乳动物肠道的主要菌[25]。变形菌门是细菌中最大的一门,包括很多病原菌。在本研究中,进境仔猪肠道菌群中变形菌门丰度降低可以认为是优势的变化。LAMENDELLA等[26]通过对猪宏基因组研究发现存在与抗生素抗性和碳水化合物代谢相关的基因,表明猪肠道微生物群可能受到饲养管理的影响,因此这种情况下需要考虑饲料等饲养管理方面的影响。
在属水平,本研究发现进境后仔猪肠道内乳杆菌属丰度显著低于进境时,且传代后这种变化仍然存在;传代后(d组)普氏菌属丰度明显增多。有多项研究表明乳杆菌是肠道中潜在的有益菌种,PERAN等[27]发现乳杆菌可以降低大鼠结肠炎模型中TNF-α的表达水平来减轻炎症反应;罗伊氏乳杆菌可以改善葡聚糖硫酸钠诱导的结肠炎模型小鼠的肠道炎症并调节肠道微生物群和代谢紊乱[28]。普氏菌属是人肠道微生物三大肠型代表菌属之一,同时也是人肠道微生物的核心菌属之一,多项研究表明其与胰岛素抵抗、类风湿关节炎和高血压等疾病呈正相关[29]。断奶后普氏菌丰度增加是由于它们能够降解半纤维素例如植物性饲料中的木聚糖[26,30],通过对差异菌属的分析,我们发现饲料的因素是不可忽视的。目前我国用于配制猪饲料的能量饲料主要以玉米和豆粕为主,法国的饲料配方主要是大麦、小麦和高粱。全大麦、全小麦配制的猪饲料能够提高肉猪瘦肉率、屠宰率、改善肉质、提高肝脏重、降低十二指肠肠壁厚度[31]。但存在无法全部代替或容易导致猪腹泻等瓶颈,这点或许可以解释为什么进境前仔猪肠道内变形菌门丰度高。
代谢功能预测发现,与仔猪刚进境时相比,传代1次的d组仔猪几种代谢功能显著下调。视黄醇(维生素A)是一种脂溶性营养素[32],是许多重要的生物功能的重要组成部分,包括生殖、胚胎发育、细胞分化、生长、免疫和视力[33]。细胞色素P450蛋白代表着具有广泛重叠基质特异性的亚铁血红素蛋白家族,负责许多外源性物质和内源性物质的新陈代谢,如类固醇、脂肪酸和前列腺素。细胞色素P450蛋白在早期胚胎发育中起着关键作用,删除细胞色素P450的小鼠胚胎在妊娠早到中期死亡[34]。肽聚糖是大多数细菌细胞壁的组成成分,是细菌主要承受压力的结构,也是真核生物免疫系统的有效刺激物[35]。目前来看,这些代谢功能的下调是劣势的,提示我们在引种饲养中注意视黄醇等物质补充。
综上,本研究认为环境变化导致肠道菌群结构改变,菌属之间的消长极有可能是优良性状快速退化的关键原因。遗传和品种因素对肠道菌群结构的影响相对较小,研究中显现出饲养管理及环境变化的影响,但仍需进行进一步研究验证。
猜你喜欢
今日农业(2022年14期)2022-09-15
中国典型病例大全(2022年11期)2022-05-13
中国典型病例大全(2022年7期)2022-04-22
大众健康(2021年8期)2021-08-04
科学导报(2021年29期)2021-06-03
科海故事博览·下旬刊(2019年6期)2019-04-16
大陆桥视野·上(2017年6期)2017-09-07
百科知识(2017年10期)2017-05-19
百科知识(2017年3期)2017-03-17
