
STAR基因缺陷致先天性肾上腺皮质增生一例
2022-07-18 05:31:36赵子辰邓姗田秦杰
生殖医学杂志 2022年7期
赵子辰,邓姗,田秦杰
(中国医学科学院 北京协和医学院 北京协和医院妇产科,国家妇产疾病临床医学研究中心,北京 100730)
类脂性先天性肾上腺皮质增生症(lipoid congenital adrenal hyperplasia,LCAH)是由编码类固醇激素合成急性调节蛋白(steroidogenic acute regulatory protein,StAR)基因缺陷导致的常染色体隐性遗传疾病。在中国,LCAH是一种非常罕见的先天性肾上腺类固醇生成缺陷疾病。典型的LCAH表现为肾上腺合成3种类固醇激素(即糖皮质激素、盐皮质激素、性激素)均缺乏、促肾上腺皮质激素(adrenocorticotropic hormone,ACTH)分泌增加及肾上腺皮质增生。我们报道1例STAR新发基因缺陷致先天性肾上腺皮质增生的中国患者,旨在提高对本病的认识,减少误诊和漏诊。
病例资料
患者,14岁,社会性别为女性,因“无第二性征发育,发现染色体为46,XY 8月”入院。
患者系其母第2胎第1产,孕期无特殊。出生时女性外阴,肤色较深。6个月时因反复感冒发热伴呕吐、嗜睡、肤色较深就诊,考虑“肾上腺功能不全”。8个月时就诊北京协和医院内分泌科,发现高血钾、低血钠,血压一直正常,诊断Addison病,予激素治疗,以后规律服用氢化可的松10 mg tid、氟氢可的松50 μg qd,电解质保持正常。4岁诊断亚临床甲减,口服雷替斯;6岁因“左腹股沟疝“行手术治疗,具体不详。
12岁时因无乳房发育和无月经初潮就诊内分泌科。为进一步发现潜在的遗传疾病,取患者外周血对肾上腺疾病相关基因(肾上腺panel)进行检测,发现STAR基因突变(表1),并由Sanger测序验证了以下两个基因突变:①NM_000349(STAR):c.64+2T>G;②NM_000349(STAR):c.677T>G(p.Val226Gly)。后者为新发突变,此前未在文献中报道过。随后,对其父母的外周血进行Sanger测序,验证了患者的STAR基因c.64+2T>G变异来自父方,c.677T>G(p.V226G)变异来自母方(图1)。根据美国医学遗传学与基因组学学会(ACMG)分类等级,STARc.677T>G功能预测为“可能致病的”。

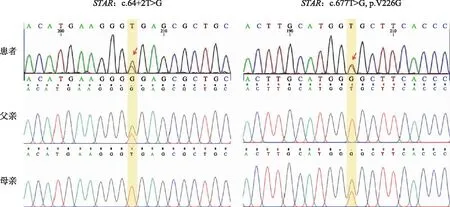
红色箭头示患者携带的STAR基因突变位点,黄色色条示患者及父母与之对应的基因位点。图1 患者携带的两个STAR基因变异及家系验证
13岁时查染色体为46,XY,激素检查结果如表2所示,促性腺激素、促肾上腺皮质激素升高,雌、孕、雄激素均降低。个人史、家族史无特殊。

表2 患者13岁时激素水平
入院查体:身高162 cm,体重76 kg,体质量指数(BMI) 28.9 kg/m2。身高年增长2 cm,近半年体重增加4.5 kg。无阴毛、腋毛,肤色偏黑,无喉结。乳房Tanner I期,外阴幼稚女性型,阴蒂不大,大小阴唇发育差,阴道可探入2 cm。
入院后完善全套类固醇激素检查,显示类固醇激素大多处于较低水平(表3)。盆腔超声未见子宫,双附件区可见中等回声,右侧2.1 cm×1.3 cm、左侧2.5 cm×1.4 cm,均未见卵泡。完善肾上腺平扫CT,提示患者双肾上腺萎缩(图2)。

表3 患者类固醇激素全套结果

A:截面1;B:截面2。图2 肾上腺平扫CT
患者婴儿期出现发热、呕吐、嗜睡、低钠高钾等肾上腺皮质功能减退症状,青春期无自主月经来潮及第二性征发育,基因检测提示STAR基因缺陷,辅助检查提示血皮质醇偏低、血ACTH升高。本患者基本符合LCAH的主要表现,家系验证支持基因缺陷遗传自父母双方。不符合之处有两点:(1)醛固酮水平为正常值低限,并无明显的降低,但由于患者长期服用氟氢可的松,不除外是外源激素补充后的水平;(2)患者肾上腺CT提示肾上腺萎缩,无明显增生。
鉴别诊断方面,常见的主要考虑是17α羟化酶缺乏症,在该疾病中,性激素和糖皮质激素的合成受影响,而盐皮质激素合成通路是正常的。17α羟化酶缺乏症的患者具有低水平的皮质醇、性激素和17-羟孕酮,而孕酮、脱氧皮质酮和皮质酮显著升高,常出现低肾素性高血压、低血钾,糖皮质激素不足的症状以及性发育异常。本患者孕酮水平很低,且血钾偏高、无高血压,同时有明确的STAR基因缺陷,不考虑17α羟化酶缺乏症。
入院后于全身麻醉下行腹腔镜探查术。术中见子宫缺如;左侧附件:左输卵管未见,可见发育不全睾丸,2.0 cm×1.2 cm×1.2 cm,其顶端可见脂肪样组织;右侧附件:右输卵管未见,可见性腺较左侧略膨大,3.0 cm×2.0 cm×2.0 cm,其顶端可见脂肪样组织(图3);乙状结肠粘连于侧盆壁。分离粘连,切除双侧性腺(图4)。
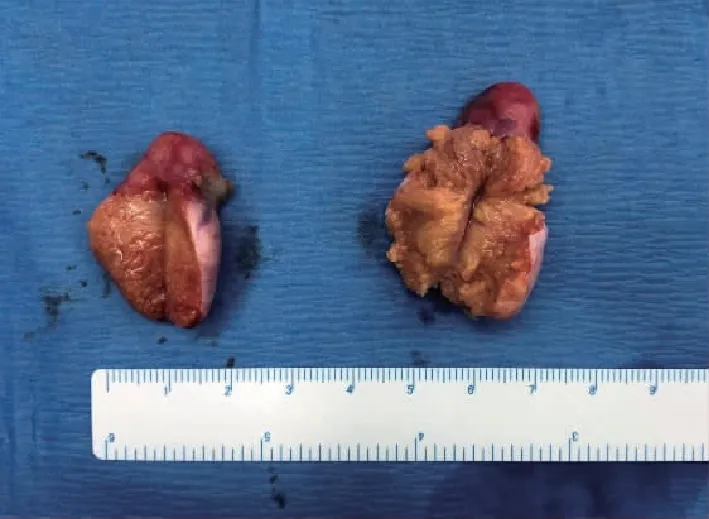
图4 切除的性腺剖面
术后病理诊断示双侧性腺可见曲细精管及附睾组织,未见生精现象。原发病方面,术后予口服激素治疗:氢化可的松tid(剂量每次分别为15 mg、10 mg、10 mg),氟氢可的松75 μg qd,补佳乐1 mg bid。亚临床甲减方面,继续予雷替斯口服75 μg qd。
患者术后两周随访,腹部切口愈合良好,继续服用氢化可的松tid(15 mg、10 mg、10 mg)、氟氢可的松75 μg qd、补佳乐1 mg bid,亚临床甲减症状给予口服雷替斯75 μg qd。术后2.5月随访,恢复良好,不易乏力、感冒,但偶尔有膝关节疼痛、腰痛,自9月初自觉乳房稍有发育,睡眠、食欲好,大小便正常。
讨 论
类固醇激素合成急性调节蛋白(steroidogenic acute regulatory protein,StAR)由STAR基因编码,它的作用是介导胆固醇从线粒体外膜向内膜的转移,这一过程为类固醇激素合成的第一步,为糖皮质激素、盐皮质激素和性激素的合成提供了底物。
STAR基因缺陷导致上述3种类固醇激素的合成不足,表现为早发的肾上腺功能不全和性腺功能衰竭。性腺发育不良在严重程度和发病时间上存在性别差异,46,XY男性患儿在胎儿期已受累,妊娠6~12周期间Leydig细胞不能合成睾酮导致男性胎儿女性化外阴,而睾丸Sertoli细胞不进行类固醇合成,其正常分泌的抗苗勒管激素抑制子宫发育,故患儿无子宫,大多数受影响的46,XY患者由于雄激素不足而缺乏男性化表现。而46,XX患者几乎均可有正常的青春期发育,在适当的年龄有月经初潮,并有可能生育后代。这可能与体内存在StAR非依赖机制的低水平甾体合成有关。46,XX胎儿卵巢不产生类固醇激素,直到青春期前卵泡细胞并未受刺激或破坏,青春期开始后LH刺激低水平的StAR非依赖性的类固醇合成,每月有一个卵泡募集并受促性腺激素刺激,导致募集细胞内胆固醇累积,细胞受胆固醇酯及其代谢物的毒性作用导致无随后的孕激素高峰发生,因此患者不排卵,表现为不孕症;而未募集的卵泡不受影响,每月一个新的募集卵泡可分泌雌激素,维持像正常月经似的周期性子宫雌激素撤退出血;患者可表现有进行性的高促性腺激素性性腺功能减退,一直到卵泡细胞被脂肪堆积而导致功能丧失为止[1]。
本例患者携带STAR基因的复合杂合突变,分别位于1号内含子与6号外显子上。1号内含子中T→G碱基的改变(c.64+2T>G)是一种剪接突变,造成转录过程中RNA前体的剪接方式改变,使产生的成熟RNA中含有内含子,造成性状的改变,是致病性的。既往文献中曾报道过两例携带这种纯合突变的患者[2]。6号外显子上T→G的点突变(c.677T>G)为错义突变,导致编码缬氨酸的密码子变成编码甘氨酸的密码子,从而使多肽链中的氨基酸序列发生改变,该突变类型此前未报道过。经家系验证,该患者的两个杂合变异分别来自父亲和母亲,位于不同的基因拷贝上,根据ACMG遗传变异分类标准预测这一新发突变(STARc.677T>G)是“可能致病的(likely pathogenic)”。ClinVar数据库中此前记录有这一位点T碱基缺失导致的移码突变(c.677del),功能预测同样是“可能致病的(likely pathogenic)”。
由STAR基因缺陷导致的LCAH疾病表现是两个独立事件的结果:STAR基因突变导致的类固醇生成缺陷是主导原因,随后肾上腺皮质中累积的胆固醇酯或进一步造成激素合成细胞的损伤。最终临床表型为婴儿期即出现的肾上腺皮质功能不全的临床症状,如高钾血症、低钠血症、低容量、酸中毒等,严重者在婴儿期死亡。患者通过盐皮质激素、糖皮质激素替代治疗可以生存到成年。皮质醇缺乏还会引起ACTH的前体阿黑皮素原的生成增加,裂解成ACTH、促黑素细胞激素,间接导致黑色素合成增加,引起色素沉着过度,因此患者的皮肤通常较黑。
大多数LCAH患者表现为双侧肾上腺增生[3]。也有个别文献报道此类患者的肾上腺是萎缩的,其中有一例中国患者的报道,患者在婴儿期就表现为早发的肾上腺危象,血皮质醇下降、ACTH升高,女性外阴而染色体为46,XY,符合经典的LCAH,唯有肾上腺是偏小的[4],与本病例类似。国外也有1例类似的肾上腺萎缩的报道,左侧肾上腺未见,右侧肾上腺缩小[5]。肾上腺萎缩的机制有待进一步研究。另有文献中报道,非经典型LCAH患者也可有肾上腺缩小的表现[6]。所谓非经典型LCAH,虽有原发性肾上腺功能不全,但性腺功能正常,同时双侧肾上腺无增生。
LCAH在中国是一种非常罕见的先天性肾上腺类固醇生成缺陷,其中完全型LCAH是最严重的类型。而在日本和韩国,这种缺乏的患病率在所有先天性肾上腺皮质增生症(CAH)类型中仅次于21-羟化酶缺乏症,男/女性别的比例是3∶1,许多受累的个体在婴儿期死亡(约有1/3靠替代治疗存活)[7]。对于存在肾上腺皮质功能减退的任何症状或体征、且外生殖器性别不清或呈女性的新生儿,应考虑该诊断;若找不到肾上腺或性腺类固醇生物合成活性的证据,则可确诊。
利益冲突声明所有作者均声明本研究不存在利益冲突。
猜你喜欢
文萃报·周五版(2022年24期)2022-06-21 20:55:40
成都医学院学报(2021年2期)2021-07-19 08:35:28
小资CHIC!ELEGANCE(2021年46期)2021-01-11 05:24:50
睿士(2020年11期)2020-11-16 02:12:27
广东医科大学学报(2020年6期)2020-02-06 06:00:58
心肺血管病杂志(2018年11期)2018-12-18 01:51:40
中华老年多器官疾病杂志(2016年8期)2016-05-14 07:16:42
食品工业科技(2014年15期)2014-03-11 18:17:46
微创泌尿外科杂志(2014年4期)2014-02-28 17:28:44
中华移植杂志(电子版)(2011年3期)2011-08-15 00:52:46
