
无定形药物固相表征技术的研究进展及应用*
2022-07-15 02:17王亚男辛俊勃
药学与临床研究 2022年3期
王亚男,李 芳,程 锦,王 咏,徐 嘉,辛俊勃,施 秦
江苏医药职业学院 药学院,盐城224005
近年来,伴随着计算化学和高通量新药筛选技术的快速发展,候选药物分子的结构越发复杂、水溶性越发差,如何有效改善候选药物的溶解度愈发成为成药性研究中需要重视的关键问题。据统计,在市售制剂中,约有40%的药物为难溶性药物,而处于研发管线中的候选药物分子中难溶性药物的比例甚至要超过70%[1]。此外,许多疗效显著的候选药物分子由于其低水溶性的问题而被迫中止于临床前研究阶段,严重限制了相关新药的开发和应用。
无定形药物,即无定形态药物,是指通过无定形化技术将药物分子从有序排列的晶态转变为长程无序排列的高能无定形态的固体形态的药物。这种晶态到无定形态的药物固态形式的改变,可有效提高难溶性药物的溶解度和溶出速率,从而显著改善难溶性药物的口服生物利用度。如用于哮喘长期治疗的Accolate®中的扎鲁司特与第二代头孢菌素类抗生素制剂Ceftin®中的头孢呋辛酯,均使用的是无定形。近年来,美国食品药品监督管理局(FDA)也陆续批准了一些由无定形药物和高分子组成的药物制剂,如2009 年批准的用于治疗由充血性心衰、肝硬化以及抗利尿激素分泌不当综合征(SIADH 综合征)导致的低钠血症的Samsca®。该制剂采用的即是喷雾干燥技术制备的托伐普坦和羟丙基纤维素的固体分散体。2014 年FDA 批准美国吉利德公司研发可用于治愈丙型肝炎的新药Sovaldi®,该制剂中使用的同样是无定形的复方雷迪帕韦/索非布韦。由此可见,对难溶性药物而言,无定形态药物制剂具有广泛的应用前景和巨大的产业价值。
药物从晶态向无定形转变需要有足够的能量来破坏晶格能,制备方法和辅料等选择均可能影响该转变的整个程度。此外,与晶态药物相比,无定形药物处在高能量的亚稳态,易在其储存和溶出过程中向其晶态形式转变。因此,在无定形态药物制剂的制备、储存和使用过程中,采用有效的技术手段对其进行表征,对解决无定形态药物制剂的物理稳定性问题极为重要。药物从晶态转变为无定形态,其物理结构、能量状态、热力学性质均发生显著改变[2]。根据无定形药物的结构和能量状态特性,有多种技术手段可对无定形药物进行鉴定分析,包括偏光显微技术、表面光栅衰减技术、X 射线衍射技术、热分析技术、振动光谱学技术、固态核磁共振技术、荧光分析技术、X射线光电子能谱(XPS)等。现对多种无定形态药物制剂研究中用到的表征技术的最新进展进行综述,并探讨其各自独特的应用,旨在为无定形态药物制剂的研究和开发提供借鉴。
1 偏光显微镜-控温热台联用技术
偏光显微镜技术是研究晶态和无定形态药物最常用的技术手段之一。与普通光学显微镜相比,偏光显微镜可将普通光转变为偏振光,从而实现鉴别某一物质是各向同性(单折射性)还是各向异性(双折射性)。晶态药物结构上各向异性,具有双折射的特性。在偏光显微镜的起偏镜和检偏镜的正交作用下,可清楚地分辨晶体药物和无定形态药物。该技术可用于无定形态药物表征、物理稳定性的初步考察以及简单的处方筛选。在实际研究应用中,偏光显微镜通常与控温热台联合使用,用于定性和定量研究在不同温度下可能发生的无定形向晶态的转变过程。Shi Q 等以抗真菌药物灰黄霉素为模型药物,采用熔融冷却法制备其无定形样品。灰黄霉素的晶体在偏光显微镜下呈现双折射现象,而无定形态灰黄霉素无双折射现象,呈现背景颜色。通过偏光显微镜和控温热台的联用,可清楚发现灰黄霉素结晶生长形貌具有明显的温度依赖性(如图1 所示)[3]。在玻璃态中(<Tg),灰黄霉素呈现的是块状结晶,接近Tg时,在灰黄霉素的结晶前沿可以看到明显的纤维状结晶,随着温度升高,灰黄霉素的晶体重新恢复块状结晶。当温度接近于药物熔点(Tm)时,灰黄霉素以单晶形式生长[3]。研究表明,这些不同的生长形貌与无定形态灰黄霉素不同温度下结晶控制机理具有差异显著相关[3]。

图1 灰黄霉素在不同温度下的结晶形貌
由于控温热台可实现精确控温,控温热台与偏光显微镜的联合使用,可被用于定量测定无定形态药物的结晶生长速率,借助该联用技术,多种反常的快速结晶生长行为被发现和报道,如玻璃态快速结晶生长,表面结晶生长以及界面诱导结晶生长[3,4]。Shi Q 等在前期研究中,通过偏光显微镜-控温热台联用技术,首次发现和报道了在过冷液体态存在的特殊的气泡、诱导快速结晶现象,并证实其结晶生长速率与此时表面结晶生长速率几乎完全一致(如图2 所示)[3,4]。近年来,偏光显微镜-控温热台联用技术还可被用于研究无定形态药物的成核[5,6]。选择通过控温热台在特定的温度放置成核,然后升温让晶核生长至偏光显微镜可见的尺寸大小,通过统计单位体积内药物的成核数量,该联用技术可定量研究药物在不同温度下成核速率及外界因素的影响与干扰[5,6]。

图2 (A)灰黄霉素130 ℃的表面结晶生长;(B)灰黄霉素130 ℃气泡诱导结晶生长;(C)结晶生长距离与时间的关系图
此外,偏光显微镜-控温热台联用技术还可为无定形态药物的制备工艺提供参考,通过分析无定形态样品在不同温度条件下的微观结构和状态,可为药物制剂的下游生产工艺如热熔融挤出技术来制备无定形态药物制剂的实验温度范围提供依据[7]。但是,目前市售的偏光显微镜的分辨率通常仅能达到微米级别,限制了其对无定形态药物及其结晶微观结构的进一步分析。此外,控温热台的升降温速率有待进一步提高,目前该联用技术对于捕捉瞬间的相变(如快速转晶等)依然存在一定的困难。
2 表面光栅衰减技术
近年来研究发现,无定形态药物在其暴露的表面可发生快速的表面结晶生长。通过偏光显微镜-控温热台技术的联用发现,表面结晶速率与体相结晶速率有着数量级的巨大差异[8]。Yu L、Huang C 等[9,10]推测,该现象极有可能与无定形药物表面快速运动的分子有关,建立了通过表面光栅衰减获取无定形态药物分子运动性的新的一种技术手段。如图3 所示,该技术首先利用控温热台将晶体药物或者物理混合物熔融并快速降温至体系Tg以下,制备成为无定形态,将温度升高至Tg以上软化无定形表面,随后将光栅压入无定形样品表面,该过程中尽量避免结晶,并降温至玻璃态移除光栅,此时无定形表面呈现规律的锯齿状结构。随后在不同温度下结合原子力显微镜或激光衍射法,表征表面锯齿状结构的高度变化的时间依赖性,并通过Mullins 方程来获取所需要的无定形药物分子表面扩散系数Ds[10]。
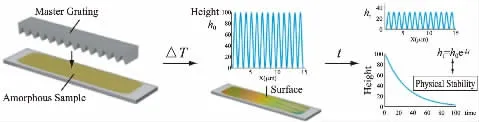
图3 表面光栅衰减技术示意图[13]

其中,K 为表面衰减常数,该常数分别与黏性流(F)、蒸发冷凝(A)、体相扩散(D)以及表面扩散(B)密切相关。方程中Ds为表面扩散系数,γ 为表面自由能,Ω 为分子体积,v 为分子数目,k 为玻尔兹曼常数。在玻璃态中,表面扩散是最重要的影响因素,而另外几个因素(Fq、Aq2和Dq3)的贡献可忽略不计。
通过表面光栅衰减技术,Yu L 等[9]成功获得了多个药物的表面扩散系数,证实无定形态药物表面发生的快速结晶现象与其表面快速运动的分子直接相关。此外,表面分子扩散速率也被证实是与分子本身的尺寸、分子间相互作用密切相关,分子尺寸越大、分子间相互作用越强,无定形态药物表面分子扩散越慢[11,12]。该技术的发展,使得人们可以从机理层面上探索无定形药物的表面结晶现象,弥补了无定形态药物在表面性质研究领域的不足。在最新的研究中,Bannow J 等[13]采用该技术研究吲哚美辛-Soloplus 固体分散体的表面分子扩散速率,随着高分子添加量的变化,证实伴随着Soloplus量增大,表面分子扩散速率逐渐变慢,此时对应体系的物理稳定性不断提高。需要注意的是,使用该技术手段获得成功的关键在于测试过程中无定形药物要避免结晶发生;但考虑到光栅衰减过程相对较为漫长,不利于维持药物的无定形态,因此,某种程度上也限制了该技术的广泛应用。此外,在多元体系中,表面光栅技术获取的是表面整体分子运动的信息;但对于如何剥离体系中不同组分材料各自的运动行为和分布则无能为力。
3 X 射线粉末衍射-同步辐射光源技术联用
X 射线衍射技术是目前国际公认的晶态物质定性与定量,以及确定无定形态最常用的分析技术。晶态药物在粉末X-射线衍射图谱中呈现为尖锐的衍射峰,而无定形态药物在粉末X 射线衍射结果则无明显的晶体衍射峰,形态上类似“馒头峰”。在无定形态药物的表征和物理稳定性测试中,常以无明显晶体衍射峰作为指标,确认无定形态药物体系制备或者贮存过程中是否发生结晶[14]。该技术同样可用于表征无定形态药物体系可能存在的微观相分离现象[15]。Newman A 等[15]以海藻糖和右旋糖酐1∶1 组成的固体分散体为对象,尽管差示量热扫描仪显示体系只有一个Tg,但通过X-射线粉末衍射技术结合分布函数耦合计算,发现体系中存在有微观的相分离行为(<30 nm)。
但需要注意的是,X-射线粉末衍射对于表征无定形态药物制剂体系中含量相对较少的结晶存在困难。近年来,伴随着同步辐射光源技术的发展,X-射线粉末衍射技术同样可以用于检测无定形态药物制剂中可能存在的痕量晶体[16-18]。Nunes C 等[16]通过同步辐射X-射线粉末衍射技术研究了无定形态蔗糖中残余的痕量晶体蔗糖,研究发现,该技术检测的含量下限可达到0.2%,远高于常规的X-射线粉末衍射技术。该联用技术高分辨率的优势主要源于同步辐射所产生的X-射线强度远高于常规的X-射线,且其准确性和稳定性均具有显著优势。同步辐射X-射线粉末衍射技术在早期的快速物理稳定性评估中具有非常重要的价值和意义。Mehta M等[17,18]结合同步辐射X-射线粉末衍射技术与宽频介电谱技术,成功构建了以水为塑化剂的无定形态药物,加速物理稳定性预测模型。虽然,同步辐射X-射线粉末衍射技术具有很好地应用前景,但同步辐射本身的光源获取需要依赖于国家同步辐射实验室等科研机构,因此一定程度上也会限制该技术的使用。
4 热分析技术
差示扫描量热分析(DSC)是热分析技术中最为常用的分析手段,可用于表征物质的结晶、熔融、玻璃化转变、晶型转变等相变过程,通过分析DSC 曲线来鉴别药物是否处于无定形态,还可用于定量检测和计算残余的晶体药物量。DSC 同样广泛用于研究无定形态药物制剂制备过程的参数控制,尤其在研究药物与高分子材料互溶性中发挥极为重要的作用[19,20]。Sun Y 等[19]以吲哚美辛和硝苯地平为模型药物,以聚醋酸乙烯酯(PVAc)、聚维酮(PVP)与聚乙烯基吡咯烷酮/醋酸乙烯共聚物PVP-VA 作为高分子载体材料,研究冷冻研磨制备的、含有不同高分子体系完全“溶解”在高分子材料所对应的温度,即结晶峰消失的临界温度,并通过公示(1)计算获得药物的“活度”a[19]。

在公式(1)中,△Hm和τm分别代表着药物的熔化焓和熔化温度;τ代表的是药物的“溶解”温度。根据Flory-Huggins理论,药物的活度与药物-高分子相互作用参数χ 又可建立如下关系[19]:
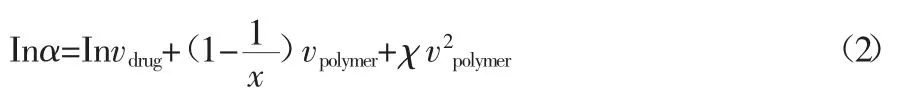
在公式(2)中,vdrug和vpolymer分别代表药物和高分子聚合物的体积分数;x 代表的则是高分子聚合物和药物的分子重量比。χ 为Flory-Huggins 相互作用参数,该参数通常可以反映药物和高分子聚合物的互溶性和相互作用[21,22]。通常该参数为负值时,意味着药物和高分子体系是热力学互溶体系[19-22],该参数可为无定形态药物制剂的处方设计、制备工艺提供参考。此外,DSC 除为常规热信号,如熔化、玻璃化转变、结晶外,最新研究表明,在无定形态药物的研究中,同样需要关注特殊的热信号[23,24]。本课题组研究发现,在低温时会出现无定形态样品脆裂的放热峰,该现象被证实与药物低温成核现象有直接联系,低温出现脆裂现象的样品被证实有极高概率发生成核,进一步支持界面诱导无定形态药物成核理论[24]。
传统的DSC 分析在测定无定形态药物Tg时,会遇到热焓、热容变化很小或多种热信号重叠的情况,此时通常会采用调制DSC 技术(MDSC)。MDSC 可有效消除动力学过程(焓弛豫和水分挥发)对无定形态药物测试的干扰和影响,更容易获得清晰和可靠的体系Tg。MDSC 对于水分和残留有机溶剂的分析能力也远优于传统DSC,特别是对喷雾干燥技术制备无定形态药物制剂的分析[25]。此外,近年来研究者也尝试DSC 技术与多种技术联合使用,如热分析-原子力显微镜联用技术(Nano-TA),就是将热分析技术和原子力显微镜(AFM)进行联合使用,可以反映样品局部微观尺度下发生的热变化,无需破坏样品即可快速获得药物的晶态或者无定形态的组成以及分布等信息[26]。Qi S 等[26]采用Nano-TA 表征非洛地平与聚维酮(PVP)形成的固体分散体薄膜中的结晶和相分离行为,Nano-TA 可明显分辨出水分导致的非洛地平-聚维酮(PVP)固体分散体的微观结构变化。Nano-TA 在表征微米以下尺寸的微观结构具有优势,但是目前该仪器造价较为昂贵,普及率并不高。
5 宽频介电弛豫谱技术
宽频介电弛豫谱技术(简称介电谱)是目前研究最广、最有可能获取无定形药物制剂中不同形式分子运动行为的弛豫时间的技术,可用于研究无定形药物制剂的整体分子运动和局部分子运动[27]。通过对样品外加交变电场,极性分子会随电场作交变的取向运动,当取消外电场时,介质分子将恢复到平均偶极矩为零的紊乱取向状态,该过程称为介电弛豫过程。如图4 所示,可通过介电谱获得不同温度条件下的弛豫谱,结合Havrililak-Negami(HN)方程获得体系的τHN[27]:

图4 灰黄霉素的介电弛豫温度依赖性图谱
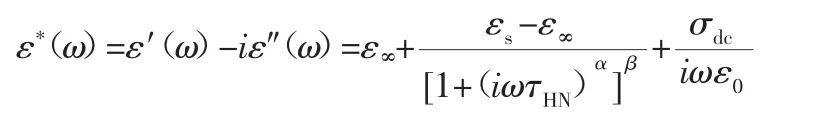
随后进而通过下式计算获得反映分子运动的弛豫时间τ[27]:
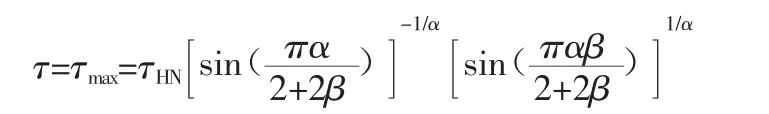
在实际无定形态药物的介电谱的研究中,通常可以同时获得反映分子整体运动初级弛豫时间(τα)的和局部分子运动的次级弛豫时间(τβ、τγ等),两者温度依赖性分别符合Vogel-Fulcher-Tammann(VFT,熔体粘度与温度的关系式)及Arrhenius 方程(由瑞典科学家阿伦尼乌斯所创立的化学反应速率常数,随温度变化关系的经验公式)[27]:

通过对这一弛豫过程的研究和分析,可获得分子运动性等信息,可与无定形药物制剂的结晶过程建立一定联系,有助于更好地预测无定形药物制剂的物理稳定性[27]。
Kothari K 等[28]在研究无定形灰黄霉素、硝苯地平以及硝苯地平-聚维酮固体分散体分子运动性时发现,介电谱获取的α-弛豫时间,可以与这些无定形态药物体系在过冷液体状态达到10%结晶度所需要的时间建立很好的相关性,该研究同时证实,该结论可以外推至其玻璃态中。此外,也有研究表明,药物和高分子聚合物形成较强的氢键相互作用时,会延长体系的α-弛豫时间(降低整体分子的运动性),此时可以更有效地抑制药物的结晶[29]。Mistry P 等[30]研究了聚维酮(PVP)、聚甲基丙烯酸羟乙酯(PHEMA)以及聚丙烯酸(PAA)3 种高分子聚合物,对无定形酮康唑分子运动性和结晶行为的影响。无定形酮康唑与3 种高分子材料分别形成弱的偶极相互作用、氢键相互作用以及离子键相互作用。其中,酮康唑与PAA 形成的离子键相互作用对其α-弛豫时间的影响最为明显,显著减低其分子运动性[30]。
Shi Q 等在国内率先探究介电谱、建立分子运动与结晶生长的相关性,在前期的研究中,以灰黄霉素和吲哚美辛为模型药物,聚环氧乙烷为模型高分子,通过介电谱研究高分子聚合物对无定形药物分子运动性的影响。对灰黄霉素研究发现,3%(w/w)聚环氧乙烷的掺杂可以使玻璃态或过冷液体态灰黄霉素结晶生长加速,达到两个数量级。此时,不同浓度聚环氧乙烷掺杂的无定形灰黄霉素的α-弛豫时间的改变与体系Tg改变密切相关,证实其对分子运动性的影响主要是“塑化作用”。但该塑化作用无法合理地解释无定形灰黄霉素的结晶加速生长行为。在随后的研究中,也证实聚环氧乙烷的掺杂会显著改变吲哚美辛部分晶型结晶动力学与流体性质的相关性,推测聚环氧乙烷的选择性加速作用与聚环氧乙烷的不同晶型表面吸附作用的强弱密切相关。因此,在通过介电谱研究无定形药物制剂分子运动与结晶动力学研究时,除需考虑整体分子运动外,还需关注高分子本身的链段动能和高分子在结晶前沿富集的现象[21,22]。此外,近年来宽频介电谱技术也在无定形药物受限态研究中取得一定进展,Zhang C 等[31]将小分子药物熔融负载入多孔阳极氧化铝材料,通过介电谱表征无定形药物受限分子运动,发现孔道尺寸和界面结构均会对无定形小分子药物的纳米结构和分子运动产生显著影响。
6 纳米红外光谱分析及拉曼光谱成像
无定形态药物与晶态药物分子的振动能量之间有一定差别,可通过红外光谱中峰型变化、峰位偏移以及峰强改变来表征。此外,红外光谱可有效表征出无定形态药物和高分子材料在固体分散体中的分子间相互作用,为无定形态药物制剂中高分子的合理筛选提供依据。近年来,纳米红外分析技术(Nano-IR)逐渐发展起来、用于表征微观尺寸下物质分布及相互作用[32]。Purohit HS 等[32]通过原子力显微镜技术(AFM)结合Nano-IR 以及Nano-TA 可检测到伊曲康唑-羟丙甲纤维素(HPMC)固体分散体中微观的相分离现象,而通过DSC 分析此时体系只有一个Tg。此外,有研究表明,傅里叶变换衰减全反射红外光谱技术(ATR-FTIR)对于无定形态药物制剂表面物质分布具有独特的优势[33]。
拉曼技术也是用于表征药物多晶型的重要手段之一[34,35],在无定形态药物制剂领域也得到了很好地应用[36,37]。拉曼散射产生的光谱图的谱带数目、位移、强度和形状,直接与分子的振动和转动信息相关联。目前,拉曼扫描成像技术成为相分离研究的关键技术之一。Tian Y 等[38]通过拉曼成像和偏光显微镜技术研究了非洛地平在醋酸羟丙基甲基纤维素琥珀酸酯(HPMCAS-HF)、聚乙烯己内酰胺-聚醋酸乙烯酯-聚乙二醇共聚物(Soluplus)和聚维酮K15(PVPK15)固体分散体中的物理稳定性和相分离行为,通过拉曼扫描成像技术,可以清楚地发现在较高载药量时,无定形态药物易于发生结晶和相分离,且相分离行为被证实与Flory-Huggins 理论预测的一致。本课题组结合偏光显微镜和拉曼扫描成像技术,证实聚环氧乙烷在灰黄霉素的结晶生长前沿出现富集现象,并通过拉曼光谱技术测定了前沿富集的高分子的含量,证实晶体-无定形态的界面高分子富集是影响无定形态二元体系结晶生长的重要因素[36]。如图5 所示,拉曼扫描成像技术同样发现吲哚美辛晶型生长过程中聚环氧乙烷具有不同的富集特征,很好地解释了聚环氧乙烷对吲哚美辛药物不同晶型的选择性及加速结晶作用[37]。此外,相比于中频和高频拉曼,低频拉曼光谱在表征分子内部振动模式更为敏感,可用于表征无定形态的形成过程。Walker G 等[39]采用低频拉曼技术清楚地展示了吲哚美辛和色氨酸在研磨过程中由晶态向无定形态转变的过程,与X-射线粉末表征技术的结果一致。需要注意的是,红外光谱和拉曼分析技术通常很少单独用于无定形药物制剂的表征,更多的是作为辅助手段和其他技术结合并相互验证。但红外光谱和拉曼技术在分析无定形药物和辅料之间相互作用时有其自己独到的优势,相关研究对于理解无定形态药物制剂物理稳定性具有非常重要的意义。图5 为吲哚美辛(LMC)不同晶型结晶生长过程、高分子聚环氧乙烷(PEO)富集现象的拉曼扫描成像[37]。
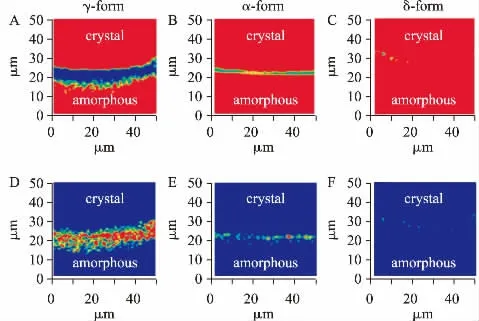
图5 吲哚美辛不同晶型结晶生长过程高分子聚环氧乙烷富集现象的拉曼扫描成像[37]
7 固态核磁共振技术
近年来,伴随着高功率偶极去偶、魔角旋转以及交叉极化等技术的发展,固态核磁共振图谱的分辨率得到显著提高,开始在无定形药物制剂研究领域崭露头角[40-44]。有研究表明,固态核磁共振技术可通过T1和T1ρ弛豫时间计算出相分离的尺寸,并据此评估无定形态药物固体分散体内药物和高分子的互溶程度[41,42]。固态核磁共振技术同样可用于表征和定量体系中不同的氢键组成[40]。Yuan X 等[40]通过13C 固态核磁共振技术研究了无定形态药物吲哚美辛的氢键组成,其中59%的氢键主要为二聚体环状结构,15%为链状结构,19%形成的是羧酸-酰胺氢键。研究指出,高分子的加入对原有氢键体系的影响同样可通过13C 固态核磁共振技术进行表征[40]。近年来,魔角自旋固体核磁共振技术被逐渐引入,应用于研究无定形药物分子间的相互作用[43]。Lu X 等[43]采用多维13C和19F 魔角自旋核磁共振技术,研究了抗真菌药物泊沙康唑从晶体转变为无定形态的分子机制。通过该技术,发现泊沙康唑晶体分子间“头-头”的分子间相互作用会在其转变为无定形后消失,但相邻药物分子间的“头-尾”相互作用则会得到保留[43]。固态核磁共振技术在无定形态药物制剂的研究中有很好的应用前景,但其普及率不高,主要受限于设备成本较高、测试时间较长、数据解析存在一定难度等诸多原因。
8 荧光分析技术
荧光分析技术是利用特殊物质在紫外光照射下产生荧光的特性及其强度进行物质定性和定量的分析方法。通过添加荧光染料或者自身具有荧光特性的药物分子,用于表征固体分散体中药物的分布、相分离以及结晶行为等[44,45]。Tian B等[44]应用吲哚美辛药物分子自身的荧光特性,结合荧光光谱分析技术,定量研究了药物分子在多个高分子基质中的相分离行为,并建立了体系互溶性与物理稳定性之间较好的相关性。Purohit HS 等[45]以伊曲康唑和羟丙甲纤维素(HPMC)作为研究对象,采用荧光成像方式研究相分离行为,采用旋转真空干燥、涂膜以及喷雾干燥3 种方法制备固体分散体,以罗丹明及氟硅酸钠为荧光剂,研究发现罗丹明主要富集在羟丙甲纤维素(HPMC)中,而氟硅酸钠主要富集在伊曲康唑中,通过荧光激发和发射光谱的差异,揭示不同制备方法下固体分散体药物和高分子材料的分布差异,为指导无定形态药物制剂的制备工艺提供依据[45]。由此可见,荧光光谱分析技术在表征固体分散体中可能存在的微观尺寸的相分离行为有很好的应用价值。近年来,荧光寿命显微镜技术也被逐步用于研究无定形药物的结晶,该技术根据药物无定形态及不同晶型的荧光衰减曲线,从而剥离出结晶信号,实现定量研究无定形态药物结晶动力学的目的[46]。但这一技术本身需要药物有荧光的性质,因此可使用的范围也相对有限。
9 X 射线光电子能谱(XPS)
XPS 通过X 射线辐射样品,激发原子或分子的内层电子或价电子,被光子激发出来的电子称为光电子。通过测量光电子的能量并做出光电子能谱图,从而获得样品有关信息。它能准确地测量原子的内层电子束缚能及化学位移,不但能提供分子结构和原子价态方面的信息,还能为电子材料研究提供各种化合物的元素组成和含量、化学状态、分子结构、化学键方面的信息。XPS 分析元素的组成和含量被用于分析固体分散体中无定形态药物的分布。Chen Z 等[47]使用XPS 技术成功地表征了喷雾干燥技术制备的固体分散体颗粒表面的药物和高分子材料分布,并以此技术为基础,深入阐明在喷雾干燥技术中影响药物和高分子表面分布的主要影响参数;该研究指出,在样品干燥的过程中,药物和高分子材料本身在溶剂中的表面张力差异、溶剂挥发扩散速率差异以及药物-高分子之间的相互作用,均会显著影响药物和高分子材料的表面分布[47]。近年研究发现,XPS 技术可与飞行时间二次离子质谱(ToF-SIMS)联合使用,这两种技术的联用可获得更高的表面分析灵敏度和空间分辨率[48]。
10 小结与展望
无定形态药物在提高难溶性药物溶解度、增加溶出以及提高生物利用度方面,具有极为重要的研究和应用价值。在无定形态药物制剂的处方开发及工业化生产中,如何通过简单有效的方法,表征无定形态药物依然是制剂领域需要重点关注的内容和重要的研究方向。此外,辅料和制备方法的选择不仅会影响制备过程中晶体向无定形态的转变,同样会影响贮存过程中无定形态药物制剂的物理稳定性。因此,如何通过有效的表征手段区分无定形态以及晶态、监测甚至是预测无定形态药物的稳定性依然是制药领域研究的重点。不同技术的灵敏度和分辨率各有差异,往往需要联合使用,而新技术的应用也将有利于加深对无定形态药物的理论认识和实践指导。相信伴随着新的检测理论和技术的发展,无定形态药物制剂表征技术的精密度和准确度均会逐步提高,并最终为无定形态药物制剂的质量控制提供更好的支撑。
猜你喜欢
纺织学报(2022年3期)2022-03-28
疯狂英语·新读写(2021年5期)2021-11-23
金桥(2020年10期)2020-11-26
中学生数理化·高三版(2016年12期)2017-03-02
电子技术与软件工程(2016年24期)2017-02-23
求知导刊(2016年27期)2016-11-07
食品与生活(2016年5期)2016-05-23
中学生数理化·八年级物理人教版(2016年9期)2016-05-14
中学生天地(B版)(2015年6期)2015-06-29
企业文化·中旬刊(2015年4期)2015-05-07
