
枸橼酸莫沙必利片质量标准中含量及其均匀度检测方法修订探讨
2022-07-12 05:20:54仝桂平毕天琛杨国宁蔡文利
中国药物经济学 2022年6期
仝桂平 毕天琛 杨国宁 蔡文利
枸橼酸莫沙必利片是一种新型胃动力药,主要用于功能性消化不良伴有的胃灼热、嗳气、恶心呕吐、早饱、上腹胀痛等消化道症状,现行标准的含量及含量均匀度采用紫外分光光度法测定,收载于《国家药品标准》新药转正标准第43册[1]。为贯彻国家对药品检验标准严谨的要求,笔者在以往的检验工作中发现,紫外分光光度法测定所用溶剂操作过程中易挥发,结果存在不稳定性。参考有关文献[2-5],经过试验,本文建立了枸橼酸莫沙必利片的含量及含量均匀度测定的高效液相色谱法(HPLC),并与紫外分光光度法进行了比较,方法验证结果表明,建立的方法准确可靠,专属性强,可为该药品质量标准修订提供参考。
1 仪器与试剂
1.1 仪器
XS105型电子分析天平(瑞士梅特勒-托利多,精密度0.01 mg),Agilent1260高效液相色谱仪(美国安捷伦),FRQ-1010T型号超声波清洗仪(杭州法兰特超声波科技有限公司),Advantage A10超纯水仪(德国默克密理博)。
1.2 试药与试剂
枸橼酸莫沙必利对照品(中国食品药品检定研究院,批号:100656-20193,按C21H25ClFN3O3·C6H8O7计,含量94.2%);枸橼酸莫沙必利片(规格:5 mg/片,企业A批号:25201212、25201207,企业B批号:124191004、124191013,企业C批号:200506、210408);枸橼酸钠为分析纯,水为超纯水,乙腈为色谱纯。
2 方法与结果
2.1 色谱条件
色谱柱:Agilent ZORBAX SB-C18(250 mm×4.6 mm,5 μm);流动相:枸橼酸盐溶液(取枸橼酸钠8.82 g,加水至1 000 ml,用稀盐酸调节pH值至4.0)-乙腈(60∶40);流速:1.0 ml/min;检测波长:274 nm;柱温:30 ℃;进样量:20 μl。
2.2 溶液制备
2.2.1 对照品溶液 精密称取枸橼酸莫沙必利对照品0.100 3 g,置50 ml容量瓶中,加乙腈-水(40∶60)适量,超声(功率:500 W,频率:42 kHz)溶解稀释至刻度,摇匀,即得对照品贮备液;精密量取5 ml,置50 ml容量瓶中,加乙腈-水(40∶60)稀释至刻度,摇匀,作为对照品溶液。
2.2.2 供试品溶液 取供试品20片,精密称定,研细,精密称取(约相当于枸橼酸莫沙必利10 mg)适量,置50 ml容量瓶中,加乙腈-水(40∶60)适量,超声(功率:500 W,频率:42 kHz)溶解并定容,摇匀,滤过,作为供试品溶液。
2.2.3 阴性溶液 按处方比例,同“供试品溶液”项下的方法制备不含枸橼酸莫沙必利的阴性溶液。
2.3 系统适用性
精密量取2.2.1项下对照品溶液20 μl,注入液相色谱仪,理论板数按莫沙必利峰计算应不低于5 000,主峰和相邻杂质峰的分离度大于1.5。
2.4 方法学考察
2.4.1 线性关系 精密量取2.2.1项下对照品贮备液0.1、0.2、0.5、1.0、2.0、4.0 ml,分别置10 ml量瓶中,加乙腈-水(40∶60)稀释至刻度,制成不同浓度的对照品溶液,按2.1项下色谱条件依法测定,记录峰面积。以浓度(X,mg/ml)为横坐标、峰面积(Y)为纵坐标进行线性回归分析,枸橼酸莫沙必利浓度在0.020 06~0.802 4 mg/ml内线性关系良好,得回归方程Y=29 491X+237.64(r=0.999 5,n=6)。
2.4.2 精密度 取2.2.1项下对照品溶液,按2.1项下色谱条件依法测定6次,测得枸橼酸莫沙必利峰面积的相对标准偏差(RSD)为0.22%(n=6),表明仪器进样精密度良好。
2.4.3 专属性 精密吸取2.2项下对照品溶液、供试品溶液、阴性溶液各适量,按2.1项下色谱条件进样测定,结果阴性溶液色谱图中无干扰峰,表明本方法的专属性良好。见图1。
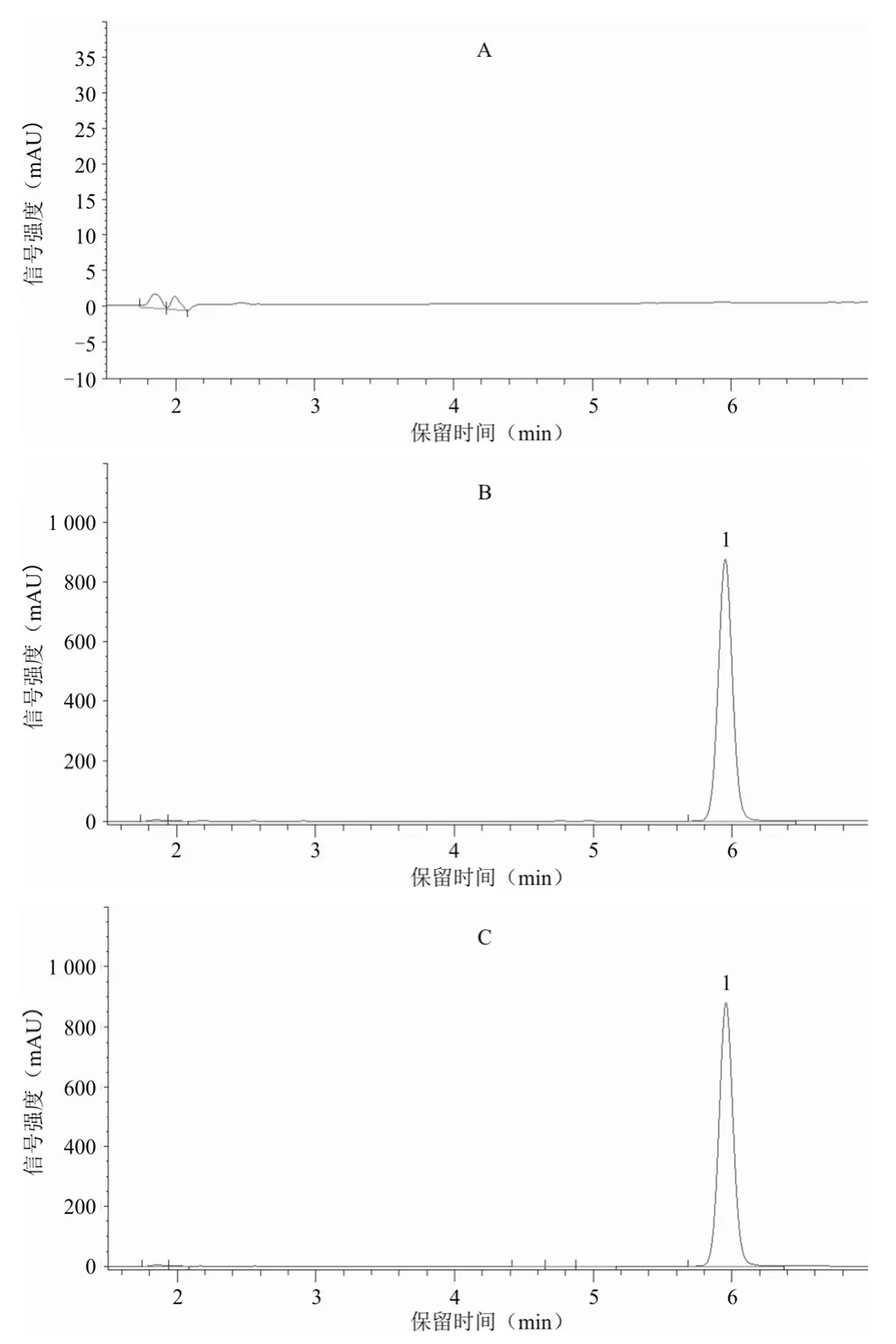
图1 高效液相色谱
2.4.4 重复性 精密称取企业A批号25201212的样品适量,按2.2.2项下方法制备6份供试品溶液,按2.1项下色谱条件依法测定,记录峰面积,计算含量。结果枸橼酸莫沙必利标示含量平均值为99.10%,RSD为0.24%(n=6)。
2.4.5 加样回收 精密称取企业A批号2520121的样品6份,分置50 ml容量瓶中,分别精密加入枸橼酸莫沙必利对照品贮备液2.5 ml,按2.2.2项下方法制备供试品溶液,2.1项下色谱条件依法测定,记录峰面积,计算回收率。平均回收率为99.02%,RSD为0.60%,表明回收率符合要求。见表1。
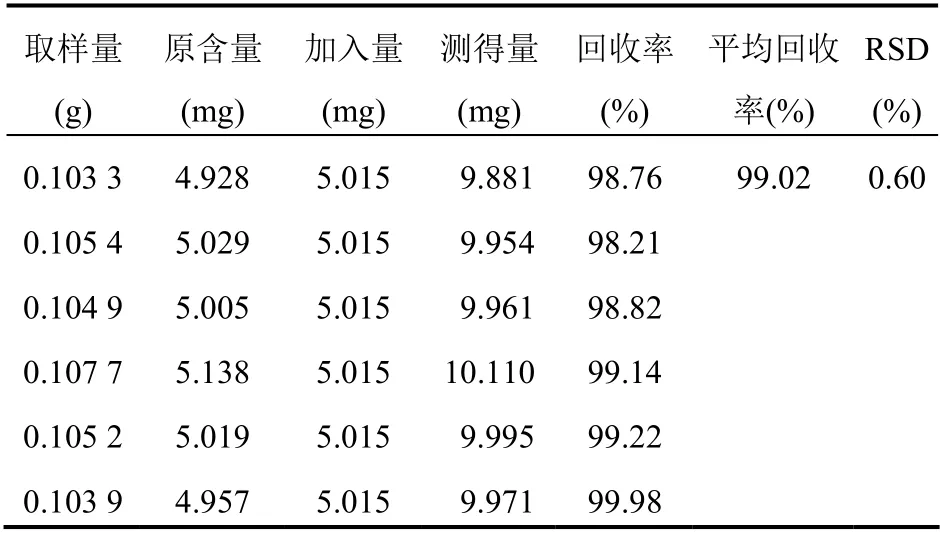
表1 枸橼酸莫沙必利加样回收试验结果(n=6)
2.4.6 稳定性 取2.2.2项下供试品溶液,分别于室温下放置0、2、4、6、8、10、12、24 h,按2.1项下色谱条件依法测定,记录峰面积。结果枸橼酸莫沙必利峰面积的RSD为0.28%(n=8),表明供试品溶液在24 h内稳定。
2.5 检出限与定量限
取2.2.1项下对照品溶液,加乙腈-水(40∶60)逐步稀释,以10倍信噪比与3倍信噪比计算定量限与检测限,结果分别为0.2 μg/ml、0.1 μg/ml。
2.6 样品含量及含量均匀度测定
含量测定:分别取1.2项下提供的6批枸橼酸莫沙必利片,按2.2.2项下方法制备供试品溶液,按2.1项下色谱条件依法测定,按外标法以峰面积计算无水枸橼酸莫沙必利的标示量,所得结果与紫外分光光度法的含量结果进行比较。两种方法的含量测定结果无明显差异。见表2。
含量均匀度测定:取1.2项下提供的6批枸橼酸莫沙必利片,每批10片,分别置25 ml量瓶中,加乙腈-水(40∶60)适量,超声(功率:500 W,频率:42 kHz)溶解并稀释至刻度,摇匀,滤过;按2.1项下色谱条件依法测定,记录峰面积,计算含量,按2020年版《中华人民共和国药典》(四部)含量均匀度检查法[6],计算A+2.2S的值,所得结果与紫外分光光度法测定的含量均匀度结果进行比较。两种方法的含量均匀度结果无明显差异。见表2。

表2 两种方法枸橼酸莫沙必利含量及含量均匀度测定结果
2.7 耐用性
考察3根不同型号的色谱柱,结果表明不同型号的色谱柱对枸橼酸莫沙必利片含量测定结果无明显影响。见表3。
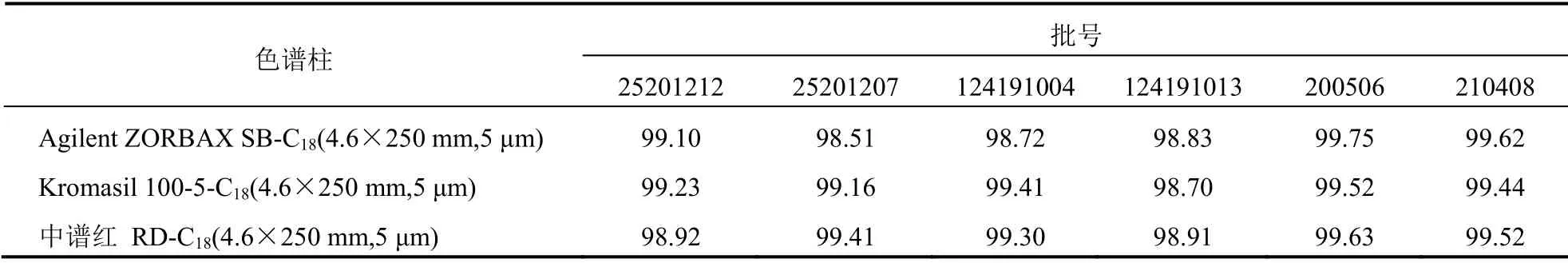
表3 耐用性试验结果(%,n=6)
3 讨论
3.1 检测波长的选择
利用DAD的在线扫描功能,在190~400 nm波长范围内对对照品溶液进行扫描。莫沙必利在225 nm、274 nm、318 nm波长处有最大吸收。225 nm波长处溶剂峰较大,310 nm波长处与主峰相邻杂质峰未检测出。因此测定波长确定为274 nm。
3.2 流动相的选择
预实验中比较了0.2%十二烷基硫酸钠的乙腈-水-甲醇-冰醋酸、0.05 mol/L枸橼酸溶液(用氢氧化钠溶液调节pH值至4.0)-乙腈、枸橼酸盐溶液(取枸橼酸钠8.82 g,加水至1 000 ml,用稀盐酸调节pH值至4.0)-乙腈、枸橼酸盐溶液(pH3.3)-乙腈等多组流动相系统,结果以枸橼酸盐溶液(pH4.0)-乙腈为流动相时,莫沙必利色谱峰对称性好,拖尾因子在0.99~1.01。考察了枸橼酸盐溶液(pH4.0)-乙腈体系3种流动相比例(50∶50、60∶40、70∶30)对测定结果的影响,综合系统适用性要求及检验时间成本,选择枸橼酸盐溶液(pH4.0)-乙腈(60∶40)作为本研究流动相。
3.3 方法的评价
将紫外分光光度法与HPLC测定的含量结果两两进行配对样品t检验(α=0.05),P值为0.744,说明两种方法测得结果无显著性差异。
紫外分光光度法采用乙醇为溶剂,在测定过程中 溶剂易挥发,稳定性差,导致含量测定结果不稳定。本研究建立的试验方法,克服了这一现象,测定结果的稳定性明显提高,该方法结果准确可靠,灵敏度高,专属性强,可为该药品质量标准修订提供参考。
(收稿日期:2022-01-27)
猜你喜欢
基层中医药(2022年7期)2022-11-17 08:25:08
中国外汇(2019年10期)2019-08-27 01:58:22
中国外汇(2019年10期)2019-08-27 01:58:22
中国外汇(2019年22期)2019-05-21 03:15:02
中国外汇(2019年21期)2019-05-21 03:04:22
医药前沿(2018年2期)2018-01-17 12:10:56
实用临床医学(2016年8期)2016-06-07 01:28:16
中国卫生标准管理(2015年18期)2016-01-20 09:27:07
中国卫生标准管理(2015年14期)2016-01-15 02:58:35
中国药业(2014年19期)2014-05-17 03:12:19
