
小儿咳喘灵颗粒微生物污染状况分析
2022-07-12 05:20刘靖华刘艳平张秀花
中国药物经济学 2022年6期
刘靖华 刘艳平 张秀花
小儿咳喘灵颗粒收载于《卫生部药品标准》中药成方制剂第四册[1],儿科用药,由麻黄、生石膏、苦杏仁、瓜蒌、板蓝根、金银花和甘草组成,配方中板蓝根、金银花等具有一定的抑菌作用[2-5],功效是润肺清热、止咳化痰,临床应用广泛。为确保临床用药安全,2016—2020年,山东省药品质量风险监测共计完成139个项目,对有关风险隐患,做到了早发现、早预警、早处置。2021年,本院继续承担小儿咳喘灵颗粒风险监测项目。微生物限度检查是评价药品安全性的重要方法,微生物污染过程涵盖生产、流通、储存、使用等环节。近年来,对原辅料、包装容器、洁净环境、生产工艺等污染的 研究越来越受到重视[6-10]。本试验首先对不同企业的样品进行微生物限度检查方法适用性试验,以消除人员、环境、试剂耗材等因素对试验结果的影响,然后根据确立的试验方法对所有样品进行微生物限度检查,对结果进行统计分析,追溯微生物污染关口,指出存在的风险隐患,为药品监管工作提供技术支撑。
1 材料与方法
1.1 材料
1.1.1 仪器 电子天平(HZT-A1000,福州华志科学仪器有限公司)、立式压力蒸气灭菌器(LDZX-50KBS,上海申安医疗器械厂)、生物安全柜(山东博科生物产业有限公司)、细菌培养箱(KB240,德国BINDER)、霉菌培养箱(MJ-250F-I,上海一恒科学仪器有限公司)。
1.1.2 培养基 胰酪大豆胨琼脂培养基,批号210829;沙氏葡萄糖琼脂培养基,批号210620;胰酪大豆胨液体培养基,批号210622;均由北京陆桥技术股份有限公司提供。
1.1.3 稀释剂 pH7.0氯化钠-蛋白胨缓冲液,批号20210624,由北京陆桥技术股份有限公司提供。
1.1.4 菌种 金黄色葡萄球菌[CMCC(B)26003]、铜绿假单胞菌[CMCC(B)10104]、枯草芽孢杆菌[CMCC(B)63501]、黑曲霉[CMCC(F)98003]、白色念珠菌[CMCC(F)98001]、大肠埃希菌[CMCC(B)44102]均由中国食品药品检定研究院生物制品检定所提供,试验所用菌株均为第4代。
1.1.5 样品 小儿咳喘灵颗粒来源分布见表1。
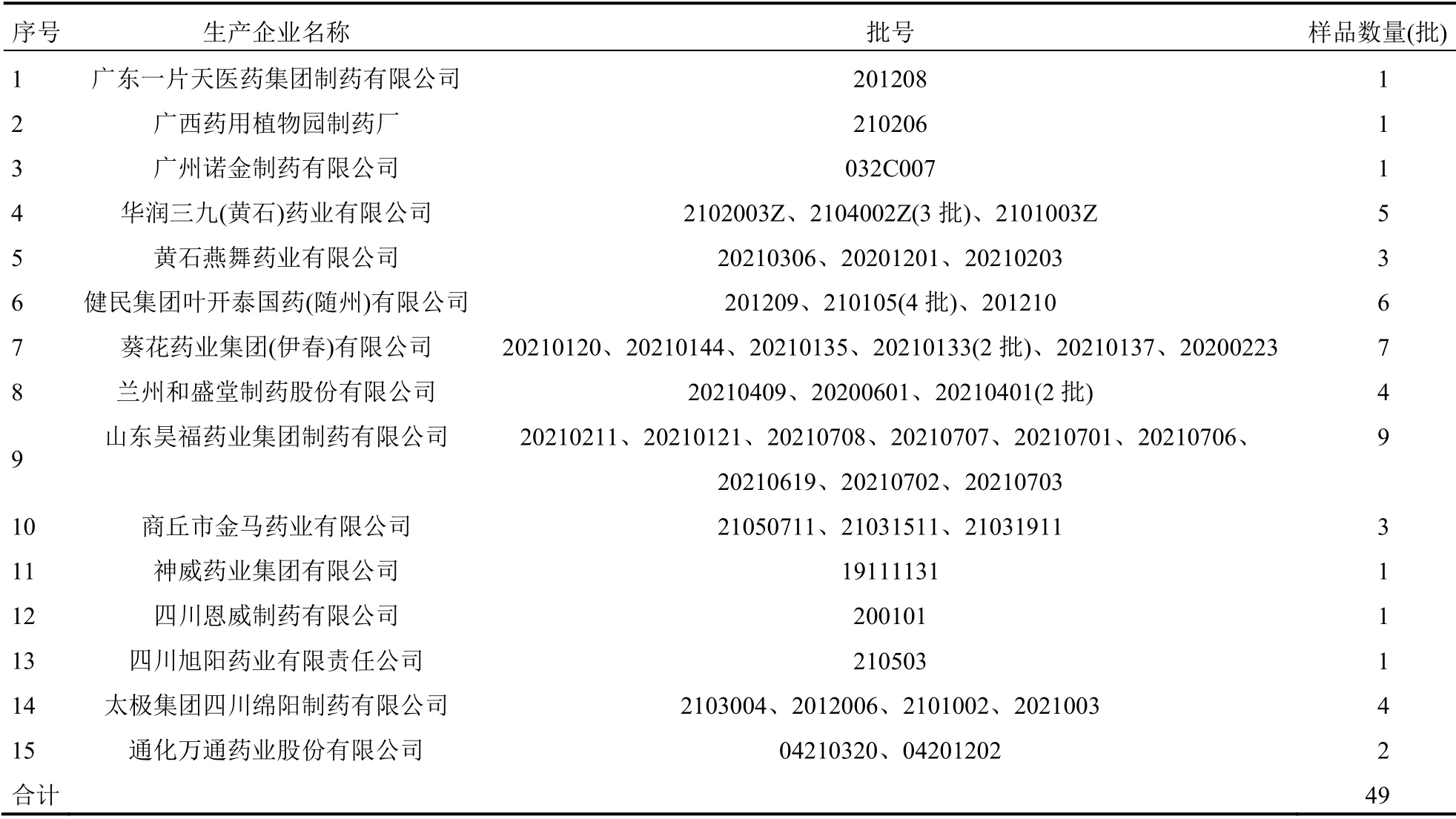
表1 小儿咳喘灵颗粒来源分布
1.2 方法
1.2.1 微生物计数方法适用性试验
1.2.1.1 菌液制备 将1.1.4项下菌种,分别划线接种于胰酪大豆胨琼脂培养基或沙氏葡萄糖琼脂培养基,按规定条件[6]培养、冲洗、稀释,制成浓度为104cfu/ml的菌液备用。
1.2.1.2 供试液制备 分别取1.1.5项下样品10 g, 加1.1.3项下稀释剂至100 ml,混匀,制成1∶10供试液备用。
1.2.1.3 微生物计数试验 试验组:分别取1.2.1.1项下菌液0.1 ml与1.2.1.2项下供试液9.9 ml,混匀备用;供试品对照组:分别取1.1.3项下稀释剂0.1 ml与1.2.1.2项下供试液9.9 ml,混匀备用;菌液对照组:分别取1.2.1.1项下菌液0.1 ml与1.1.3项下稀释剂9.9 ml,混匀备用;以上3组分别取1 ml注入平皿中,按照《中华人民共和国药典》2020年版四部通则1105[11],加入相应的胰酪大豆胨琼脂培养基或沙氏葡萄糖琼脂培养基,培养、计数,计算各试验菌回收率。
1.2.2 控制菌检查试验 取1.2.1.2项下供试液10 ml至100 ml胰酪大豆胨液体培养基中,以大肠埃希菌为阳性对照,胰酪大豆胨液体培养基作为阴性对照,照《中华人民共和国药典》2020年版四部通则1106[12],培养、选择和分离培养、结果判断。
1.2.3 结果判断 计数方法适用性试验中,试验组菌落数减去供试品对照组菌落数的值与菌液对照组菌落数的比值应在0.5~2范围内[11]。
2 结果
2.1 微生物计数
由表2可以看出,各试验菌回收比值均在0.5~2范围内,符合试验要求,该方法可用于微生物计数检查。
2.2 控制菌检查
由表3可以看出,阳性对照呈阳性,阴性对照呈阴性,符合试验要求,该方法可用于控制菌检查。
2.3 样品检查
由表4可以看出,49批样品需氧菌总数均小于103cfu/g,检出率为30.6%,霉菌和酵母菌总数均小于102cfu/g,检出率为20.4%,大肠埃希菌(1 g)均未检出。

表4 49批样品微生物限度检查结果
3 讨论
本次风险排查抽验任务,样品分布广泛,生产环节涉及全国15家企业,经营环节涉及全省33家企业。因原料产地、批次、辅料及生产工艺存在一定差异,不同企业的方法适用性试验不同[13]。针对这一问题,首先采用常规法分别对15家生产企业的样品进行微生物限度方法适用性试验。从表2可以看出,15家生产企业微生物计数各试验菌回收比值均在0.5~2范围内;从表3可以看出,控制菌检查阳性对照组均呈阳性,阴性对照组均呈阴性。结果表明,49批样品均可采用常规法进行微生物限度检查。

表2 微生物计数方法适用性试验各试验菌平均回收比值(n=3)

表3 控制菌检查方法适用性试验结果
微生物限度检查是非无菌制剂质量控制的重要方面,也是保障制剂安全的重要措施[14]。从表4可以看出,49批样品中需氧菌总数小于10 cfu/g共34批次,10~100 cfu/g共14批次,101~200 cfu/g共1批次,检出率为30.6%;霉菌和酵母菌总数小于10 cfu/g共39批次,10~50 cfu/g共6批次,51~100 cfu/g共4批次,检出率为20.4%;控制菌均未检出。结果符合规定[15]。从检出率来看,微生物计数的检出率在20%~30%,说明微生物污染的概率并不高,尤其是霉菌和酵母菌。49批样品在人、机、料、法、环等试验条件完全一致的情况下,个别数据出现异常,完全可以排除本实验室对检验结果的影响。研究发现,仅霉菌和酵母菌总数在90~100 cfu/g有3批,结果为临界值,与标准限值102cfu/g相近,说明样品污染的因素可能与原辅料产地、储存条件、生产环境等密切相关。进一步研究发现,3批样品涉及2家生产企业,且有1家生产企业的2批样品为关联批号,说明样品污染的主要原因很可能是原辅料在购进或储存过程中受到微生物污染。《中华人民共和国药典》2010版[16]规定,不含药材原粉的口服给药制剂霉菌和酵母菌数不得超过100 cfu/g,而《中华人民共和国药典》2015版[17]及2020版[15]规定,霉菌和酵母菌总数为102cfu/g,可接受的最大菌数为200 cfu/g;从标准的修订过程中可以看出,对微生物污染的控制越来越受重视,是确保药品质量安全性及有效性的第一关口,相关生产企业须引起高度重视。同时,药品监督管理机构,必须严格按照药品生产质量管理规范、药品经营质量管理规范及药品管理法的要求,加大监管力度,提高监管效能,保障人民群众安全用药。
猜你喜欢
数学小灵通(1-2年级)(2021年11期)2021-12-02
中国土壤与肥料(2021年5期)2021-12-02
纺织科技进展(2021年4期)2021-07-22
家庭影院技术(2021年2期)2021-03-29
数字海洋与水下攻防(2020年5期)2021-01-04
中等数学(2020年8期)2020-11-26
疯狂英语·新悦读(2020年7期)2020-07-30
小学生学习指导(低年级)(2020年4期)2020-06-02
癌变·畸变·突变(2020年1期)2020-02-12
数学大王·低年级(2019年8期)2019-08-27
