
有机物辅助的硫化物电解质基固态电池
2022-07-07 03:32:00李一涛沈凯尔庞全全
储能科学与技术 2022年6期
李一涛,沈凯尔,庞全全
(北京大学材料科学与工程学院,北京大学先进电池材料理论与技术北京市重点实验室,北京100871)
如今锂离子电池(lithium-ion batteries,LIBs)已经成为了最普遍的可商用化学储能器件之一,应用场景从手机等电子产品到电动汽车[1-2]。而随着电动汽车和电网储能需求的快速增长,迫切需要高能量密度、高安全性的电池[3]。为了使电池达到更高的能量密度,锂(Li)金属由于其极高的理论比容量(约3860 mAh/g)、低密度(0.59 g/cm)和最低的负电化学电位(-3.04 Vvs.标准氢电极)成为了一种理想型的负极材料[4-5]。然而传统的LIBs 使用的是低闪点、易燃的碳酸酯类溶剂(EC、DMC 等)作为电解液,其中锂金属在负极上不稳定地沉积/剥离会形成枝晶,枝晶的生长不仅会严重影响电化学性能,还会刺穿隔膜导致电池内部短路,最终引发着火等严重的安全问题[6-7]。利用具有较高机械强度和热稳定性的固态电解质(solid-state electrolytes,SSEs)作为易燃电解液的替代,是目前科研人员和电池业界提升电池安全性的有效策略[8-9]。
目前人们已经开发出众多种类的固态电解质:聚合物电解质(polymer electrolytes)、氧化物基固态电解质(oxide-based SSEs)、硫化物基固态电解质(sulfide-based SSEs)、卤化物基固态电解质(halide-based SSEs)等,其中氧化物SSEs通常更为“脆”和“硬”,而硫化物SSEs则更“软”,其杨氏模量比氧化物低,更容易进行加工并形成致密层[10]。在近年来的研究中,硫化物SSEs 呈现出可以与电解液媲美的室温离子电导率,包括玻璃-陶 瓷(glass-ceramic)型:Li7P3S11(70Li2S·30P2S5)(17 mS/cm,Ea=0.18 eV)[11-12];硫代-LISICON(Thio-LISICON)型:Li10GeP2S12(12 mS/cm,Ea=0.25 eV)[13]、Li9.54Si1.74P1.44S11.7Cl0.3(25 mS/cm,Ea=0.24 eV)[14];锂-硫银锗矿(Li-argyrodite)型:Li6PS5X(X = Cl,Br,I)(约1 mS/cm,Ea=0.3~0.4 eV)[15-16]、Li5.5PS4.5Cl1.5(9.4 mS/cm,Ea=0.29 eV)[17]。尽管硫化物固态电池理论上比液态电池具有更高的能量密度和安全性,但同样面临着诸多困难,例如:界面空间电荷层效应[18],较差的电极/电解质界面接触[19],对空气、水稳定性差[20]等。Liang 等[21]总结了基于硫化物SSEs 的固态电池中的一些界面问题,如图1。人们已经开发出了很多改善界面和提高硫化物SSEs 性能的方法,包括:构筑稳定的固态电解质表面保护层[22]、通过引入新组分改善空气稳定性和电导率[23]、利用溶液法合成硫化物SSEs[24-26]等,其中很多的方法都需要应用各种有机溶剂和引入有机组分来实现对硫化物SSEs 性能的增强[27]。然而由于含P 元素硫化物中P—S 键的键能远低于P—O键,从而使得P—S结构容易发生氧化或者脱硫[28],就导致实际上硫化物SSEs对有机组分的耐受要求十分苛刻,故而能够进行配合的有机组分种类和数目极其有限。
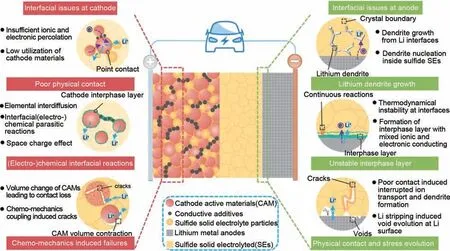
图1 硫化物SSEs的固态电池中各种界面问题示意图[21]Fig.1 Schematic diagram of various interface problems in the solid-state batteries of sulfide SSEs[21]
本文紧扣有机物(包括有机溶剂和聚合物)对硫化物SSEs配合时的增益作用,首先对近年来基于硫化物SSEs的准固态电池研究进展进行了总结与回顾;其次讨论了全固态电池中聚合物/硫化物复合固态电解质的湿法和干法制备;分类总结了有机物辅助的硫化物SSEs对全固态电池复合正极的性能改善方法;最后对有机物增强硫化物基固态电池的方式进行了总结与展望。
1 硫化物基准固态电池
通常认为在全固态电池(all-solid-state batteries)中,固态电解质与电极界面接触、正极活性物质(cathode active materials,CAMs)与其余组分接触为“点对点”(point-to-point)接触,这样就会导致在内外部的界面上存在很多空隙[21,29],而在电池中加入少量的电解液即可极大地提升正极的离子电导率,并且增加界面的接触和浸润[30]。准固态电池(quasi-solid-state batteries,QSSBs)就是在复合正极内部、电极/固态电解质界面或固态电解质内部加入少量电解液的电池,通常电解液的加入量较少,且在电池中不可自由流动。基于聚合物电解质的准固态电池已经有很多报道,其中与电解液结合的聚合物电解质多被称为凝胶聚合物电解质(gel polymer electrolyte)[31-32]。而基于硫化物SSEs的准固态电池是本综述的重点讨论内容。
1.1 准固态电池中的复合正极及其界面
相比于硫化物SSEs 的低杨氏模量,氧化物SSEs面对的复合正极、正极/电解质界面问题更为严峻[3]。鉴于出色的结构稳定性和空气稳定性,基于氧化物SSEs与离子液体(ionic liquids,ILs)的准固态电池已经有不少报道[33-36]。而在硫化物基准固态电池中,对电解液的筛选提出了两个要求:首先要求溶液不与硫化物SSEs发生化学反应;其次要求该溶液能够溶解Li 盐,并提供较高的离子电导。这样看上去两个要求存在一定的矛盾,因为硫化物SSEs本身就可以看作一种复合锂盐。Oh等[37]从锂硫(Li-S)电池高浓型电解液(high concentration electrolyts)的报道中获得灵感,利用溶剂化离子液体(solvate ionic liquids,SIL)对多硫化锂(lithium polysulfides,LiPSs)溶解的抑制作用,将三甘醇二甲醚(triglyme,G3)作为溶剂,与双三氟甲烷磺酰亚胺锂[lithium bis(trifluoromethanesulphonyl)imide,LiTFSI]以1∶1 的摩尔比例混合,制备了Li(G3)TFSI(LiG3)电解液,对硫化物SSEs(LPS、LGPS)较为稳定[图2(a)]。作者的解释是,G3溶剂中的氧(O)通常会通过亲核攻击来攻击硫化物SSEs的正电元素(P 和Ge),而G3 与Li 盐的强络合显著削弱了O 的供体能力,尤其是G3 中的4 个O 与Li 离子的强配位会降低O 的亲核性,导致LiG3 电解液的反应性显著降低[图2(b)]。并且根据软硬酸碱理论(hard and soft acids and bases theory,HSAB),硬酸倾向于与硬碱反应,软酸则与软碱反应[38]。例如,P 是比Ge 要硬的酸,所以更容易被硬碱O 亲核攻击,这也可以解释为什么LGPS 比LPS 在LiG3 中更加稳定。以LFP∶LGPS∶C∶LiG3=37.7∶50.9∶5.7∶5.7 的质量比例制备复合正极,其中LiG3 添加量约为0.3 μL/cm2,LiFePO4||Li-In电池获得了144 mAh/g(0.1 C)的第二圈放电比容量。
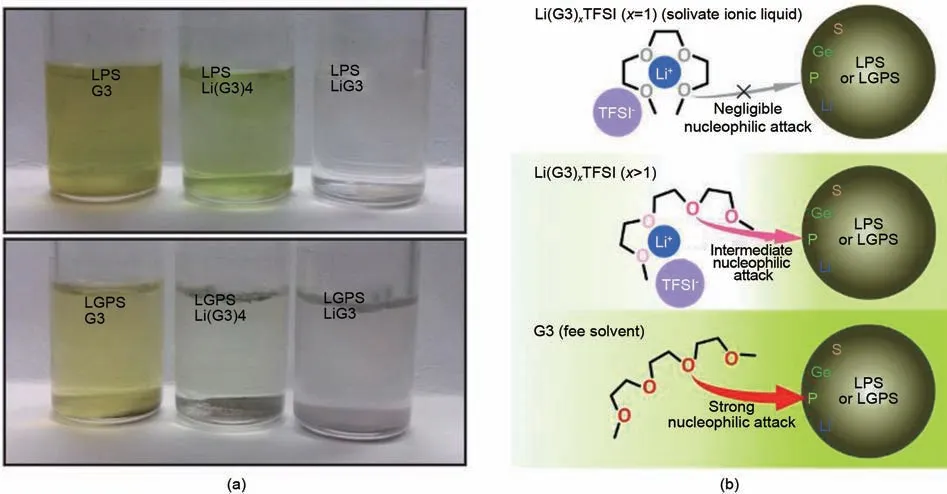
图2 (a)LPS和LGPS粉末在分别加入G3、Li(G3)4和LiG3溶液7天后的照片;(b)LiG3系列溶液与电解质的反应性示意图[37]Fig.2 (a)Photographic images of mixtures of LPS and LGPS powders with the liquids G3,Li(G3)4,and LiG3 after being kept for 7 d;(b)Schematic diagrams of the reactivity of LiG3 solution series with electrolytes[37]
在后续的研究中,Oh 等[39]通过引入丁腈橡胶(nitrile butadiene rubber,NBR)作为黏结剂,以弱极性的二溴甲烷(dibromomethane,DBM)作为溶剂,通过将混合浆料刮涂在铝箔上制备了LiNi0.6Co0.2Mn0.2O2(NCM622)复合正极,其中质量比例为NCM622∶LPSC∶C65∶NBR∶LiG3=70.0∶24.0∶1.0∶1.5∶3.5。以Li-In 为负极的电池获得了174 mAh/g(0.1 C)的初始放电比容量。作者同时指出,无极性的邻二甲苯、弱极性的DBM不仅不会破坏LiG3 的稳定溶剂化结构,也不会亲核攻击LPSC中的P,并且能够很好地溶解NBR等弱极性黏结剂。受LiG3对硫化物SSEs具有较好稳定性的启发,Cao 等[40-41]将约25 μL 的LiG3 加入复合正极之余,同时利用其湿润正、负极界面,所组装的S|LGPS|Li 和Li4Ti5O12|LGPS|Li 准固态电池均获得了不错的性能。还有的研究者将电解液的添加量提高到复合正极质量的60%,也获得了不错的全电池性能[42]。而为了获得良好的浸润效果,还可以在正极和SSEs中间引入含有电解液的玻璃纤维隔膜,并同时起到阻隔的作用[43]。由此可见添加电解液到复合正极的内部及界面处,对改善电池性能确实具有一定的积极作用。
1.2 准固态电池中的固液混合电解质
如前文所述,由于硫化物SSEs 对溶剂和空气较差的稳定性,使得在硫化物SSEs中直接添加电解液以提高性能的效果变得十分困难,并需经过很长时间的探索。早在2010 年,Minami 等[44]就试图在75Li2S-25P2S5玻璃SSE中加入不同种类的ILs来促进其离子电导率和拓宽电化学窗口。通过将硫化物SSE 前驱体与10%(物质的量分数)EMIFSA[1-ethyl-3-methyl imidazolium bis(fluorosulfonyl)amide]以及0.3 mol/kg LiTFSA共同球磨,可以得到室温离子电导率约10-3S/cm的复合电解质。随后,同一研究团队的Shimamoto 等分别探究了羧酸酯(Carboxylate esters)类电解液[45]和磷酸酯(Phosphate esters)类电解液[46]对玻璃型硫化物SSE成型性能的促进作用。但上述工作都只停留在复合电解质的制备与性能表征阶段,尚未组装可稳定运行的QSSBs。并且即使加入适量电解液改善了硫化物SSEs 的成型性能、降低了界面阻抗,但也会带来不容忽视的离子电导率损失[37]。Tan 等[47]将Li7P3S11电解质分别加入H2O、NMP、DMF、乙腈、甲苯、二甲苯等溶剂中,通过观察电解质沉淀物晶体结构是否发生变化来判断溶剂对Li7P3S11的影响。作者发现,当分散在具有高介电常数的极性溶剂,如乙腈和碳酸二甲酯(DMC)中时,电解质发生化学降解;而当使用低介电常数的非极性溶剂,如甲苯和二甲苯时,化学结构和离子电导率得以较好保留。
同样受到Li-S电池微溶型电解液的启发,固液混合准固态电解质的研究者们逐渐将目光转向含有氟代醚(HFE)的电解液体系。例如Philip 等[48]基于1,3-二氧戊环(1,3-dioxolane,DOL)、乙二醇二甲醚(1,2-dimethoxyethane,DME)和氢氟醚(1,1,2,2-tetrafluoroethyl 2,2,3,3-tetrafluoropropyl ether,TTE)配制了7 mol/L LiTFSI DOL/DME 电解液,TTE 用于稀释。50 μL 的电解液被滴加在了LGPS电解质冷压成型的圆片两侧。LGPS相对LPS电解质显示出了在该电解液中较低的溶解度,并且没有在表面形成钝化层。Li||Li对称电池显示出准固态电解质具有更低的界面电阻和更好的循环稳定性。Shin等[49]配制了一种基于乙腈(acetonitrile,MeCN)和TTE 的局部高浓电解液(MeCN)2-LiTFSI∶TTE,将40%(质量分数)的电解液与60%(质量分数)的Li7P3S11电解质进行预混合,并组装扣式Li2S||Li-In准固态电池进行测试。固液混合的电解质首先改善了正极的浸润性和界面接触,并实现了88%的活性物质利用率;其次由于电解质/电极界面不像传统的固态电池那样坚硬,电解质层可以适应体积变化,减小了界面处应力/应变引起的界面阻力,同时抑制了裂纹的形成。组装的电池在80 圈后仍保持了880 mAh/g(0.1 C)的高放电比容量和较长的循环寿命。除此之外,纯的HFE 溶剂也被用于与硫化物SSEs混合,并组装QSSBs进行测试[50]。由于基于硫化物SSEs的固液混合电解质还处于较为初始的探究阶段,电解液的添加量通常较多,并且对大部分硫化物SSEs与溶液接触后自身性质的变化细节和机理也没有系统完善的讨论,这一部分的工作仍然需要不断地推进。
1.3 准固态电池中的负极及其界面
由于固态电池多使用金属Li作为负极,而Li在电池循环过程中又会饱受Li枝晶的困扰,因此在组装电池时对负极界面进行修饰成为了很重要的手段[3,10]。目前常见的负极保护法包括:①使用金属合金负极,例如Li-In 合金[51];②插入对Li 金属和硫化物SSEs 都化学稳定的电解质或无机层[52-53];③在Li金属表面原位形成一层钝化保护层[54]。而在形成人工SEI 保护层之余还能降低界面阻抗的方法,就是组装含有电解液的QSSBs。通常使用含F的溶剂来实现这一策略。
Xu等[50]利用一种甲基九氟丁醚(Methoxyperfluorobutane)在150 ℃的条件下处理Li金属,在Li表面形成了一层致密的LiF人工SEI层,降低了界面阻抗。由于LiF对硫化物SSE和Li都是电化学稳定的,且具有较低的表面能,与HFE混合电解质配合能够抑制锂枝晶的生长。值得一提的是,作者同时还使用了固液混合的准固态电解质,将120 mg 的Li7P3S11电解质与约0.1 mL的HFE 进行混合,并以铌酸锂包覆的钴酸锂(LNO@LCO)为正极组装全电池,初始放电比容量达到118.9 mAh/g(0.1 mA/cm2)[图3(a)]。Wan 等[55]配 制 了1 mol/L LiTFSI-Mg(TFSI)2-DME(Li∶Mg=2∶2,摩尔比)电解液,在组装全电池时将约20 μL 电解液滴加在LGPS 和Li 金属的界面。由于憎锂性的差异,LiTFSI-Mg(TFSI)2先在Li 表面形成亲锂的LixMg 合金层,并在LixMg顶部形成憎锂的LiF 层。最后还原的DME 在LiF 层和LGPS之间形成有机组分富集层[图3(b)]。除了降低了界面阻抗之外,进行界面处理后的Li|LGPS|Li对称电池的临界电流密度可达1.3 mA/cm2。组装的Ni-Li2S-LiTiS2/LGPS-LiMg22/Li 全电池具有699.7 mAh/g(100 mA/g)的初始放电比容量。
而同一研究小组的Zheng[42]和Umeshbabu[56]分别使用1.5 mol/L LiTFSI|Pyr13TFSI 和1 mol/L LiTFSI|Pyr13TFSI[N-propyl-N-methyl pyrrolidinium bis(trifluoromethanesulfonyl)imide]离子液体滴加在Li金属表面,原位形成人工SEI层。SEI层的主要成分是LiF,并且在电池循环之前就可以形成薄层并将硫化物SSE与Li金属隔开以阻止副反应的发生。从组装的Li||Li 对称电池随时间变化的阻抗图来看[图3(c)]:随着时间的增加,体电阻Rbulk值几乎是恒定的,这表明硫化物SSE的离子电导率不会随着存储时间的增加而改变。然而界面电荷转移电阻Rintf有显著变化:首先在前3天随时间急剧上升,最终可以在15 天内保持不变,这意味着由于添加离子液体,硫化物SSE和Li金属电极之间形成了稳定的界面。电阻的初始增加是由于钝化层的生长,它随着时间的增加而变得稳定。所组装的LiFePO4|Li10SnP2S12|Li电池和S|LGPS|Li电池分别可达到144 mAh/g(0.1 C)和1068 mAh/g(0.05 C)的初始放电比容量。相似的策略也被使用到了Na 金属QSSEs 中,Li 等[57]使 用0.2 mol/L NaTFSI|Pyr14TFSI[1-butyl-1-methyl pyrrolidinium bis(trifluoromethanesulfonyl)imide] 和Na3SbS4电解质组装了FeS2||Na全电池,Na金属界面得到了稳定,并且在循环30圈后达到了300 mAh/g(20 mA/g)的放电比容量。
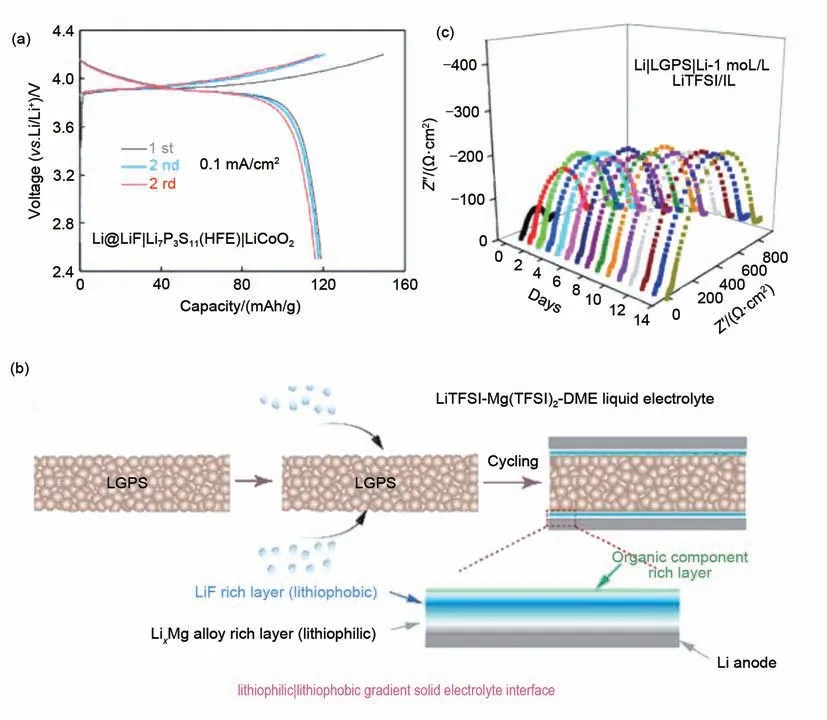
图3 (a)Li@LiF|LPS(HFE)|LCO电池在室温下电流密度为0.1 mA/cm2时的充放电曲线[50];(b)在LGPS膜表面滴加电解液后,Li和LGPS之间LixMg/LiF固体电解质界面的原位形成示意图[55];(c)加入1 mol/L LiTFSI/IL的Li|LGPS|Li电池在不同存放时间后随时间变化的阻抗图[56]Fig.3 (a)Charge-discharge profiles of the Li@LiF|LPS(HFE)|LCO cell at current density of 0.1 mA/cm2 at room temperature[50];(b)Schematic diagram of in situ formation of the LixMg/LiF solid electrolyte interphase between Li and LGPS after dropping liquid electrolyte onto the LGPS membrane surface[55];(c)Time evolution of impedance response of the Li|LGPS|Li cell with 1 mol/L LiTFSI/IL at various storage times[56]
有别于上文提到的对Li金属负极界面采取的修饰手段,为了从源头上阻止枝晶的产生,近几年来碱金属基有机液态金属溶液(例如:Li-Na-K-Bp-DME)负极也成为了一个新颖的研究方向,并已经应 用 于Na-S 电 池[58]、K-O2电 池[59]、Li-O2电 池[60]。Peng 等[61]将Li 金属溶解在联苯(biphenyl,Bp)和DME溶液中,制备了Li1.5Bp3(DME)10液态负极,在室温下具有12.2 mS/cm 的总电导率。其独特的性质在于作为一种溶剂化电子溶液,固态的Li金属不会在循环中出现,而是由Li+与联苯自由基阴离子形成稳定的[(Bp·-)Li+](C4H10O2)结构,如图4(a)。为了防止液态金属负极扩散进入Li7P3S11电解质引起短路,作者使用玻璃纤维容纳液态负极,并在电解质表面覆盖了聚氧化乙烯(PEO)层,然后组装Li||Li对称电池进行测试,见图4(b)。带有PEO 保护层的电池具有较低的初始过电位(0.11 V)和较好的循环稳定性(≈3000 h),并且能够达到17.78 mA/cm2的临界电流密度[图4(c)]。
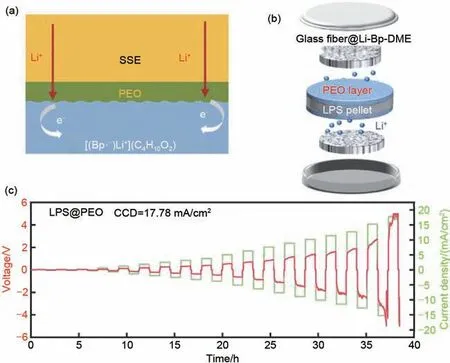
图4 (a)Li-Bp-DME液态负极与PEO保护的硫化物SSE形成的溶剂化结构示意图;(b)用于CCD测试的对称扣式电池示意图;(c)采用PEO保护层的对称电池在阶梯电流密度下的电压分布[61]Fig.4 (a)Schematic diagram of solvate structure in battery with Li-Bp-DME liquid anode with PEO protective sulfide SSE;(b)Schematic illustration of the symmetric coin cell used for CCD detection;(c)Voltage profiles of the symmetric cell using PEO protective layer at step-increased current densities[61]
需要特别指出的是,在准固态电池的开发过程中,仍然不应该放松“高安全性”这一固态电池的重要目的。虽然目前的研究中电解液的加入可以稳定硫化物SSEs并降低界面阻抗,但大量易燃有机溶剂的使用反而增加了电池的安全隐患,乃至降低了电芯的能量密度,所以电解液的添加策略仍然需要经过慎重选择与控制。
2 全固态电池中的聚合物/硫化物复合电解质
为了在实现SSEs 高离子电导率的同时还具备更好的可加工性和柔性,研究者们将无机电解质与聚合物电解质结合起来组成复合电解质。其中面临的主要挑战是,当在无机电解质中加入离子绝缘的聚合物时,要保持无机部分的高离子导电性[10]。笔者将目前主流的复合电解质制备方法分为湿法和干法,并根据聚合物的极性等特点进行分类讨论。
2.1 湿法制备的复合电解质
为了提高电池的能量密度和SSEs 的柔性,就需要将硫化物SSEs 从传统的冷压成块状(pellettype)转向可大面积制备的薄片状(sheet-type)或薄膜(membrane)状。其中湿法浆料制备作为传统电池电极制备的技术,已经在制备sheet-type电解质中逐渐成为主流。在复合的过程中,聚合物起到的作用也不尽相同,有的是作为柔性的导电网络,有的作为黏结剂,还有的仅作为支撑的骨架材料。研究者们参考聚合物加Li 盐的固态聚合物电解质(solid polymer electrolytes,SPEs)制备方式,将硫化物SSEs 与传统的具有较强极性的聚合物(例如PEO、PVDF)共同溶于有机溶剂来制备复合电解质。在早期阶段,硫化物SSEs 更多是被当做活性填料填充进入聚合物的网络中,起到降低聚合物结晶度,促进离子电导率的作用[62]。其中硫化物SSEs 的添加量通常不超过电解质总质量分数的50%,所用的溶剂也多为乙腈、四氢呋喃(THF)等极性较高的溶剂,总体的离子电导率体现的是聚合物的性质,很难在室温达到10-4S/cm[63-65]。
而在硫化物SSEs 含量较多的传统极性聚合物复合体系中,离子电导率的损失也仍然不容忽视。Wang 等[66]将95%(质量分数,下同)的LPSC 与5%的PVDF 溶于乙酸乙酯(EAC)中,通过蒸发溶剂得到约120 μm厚度的电解质膜。复合电解质的室温离子电导率为1.21×10-3S/cm(0.29 eV)。Zhang等[67]使用约95%的78Li2S-22P2S5电解质、LiTFSI 和PEO、PVDF分别溶于甲苯和乙酸乙酯,通过蒸发溶剂得到混合物粉末,并冷压粉末得到约120 μm厚度的电解质膜,室温离子电导率约4×10-4S/cm。Liu 等[68]将约90%的LGPS 与PEO、3-氯丙基三甲氧基硅烷[(3-chloropropyl)trimethoxysilane,CTMS]溶于苯甲醚中,通过刮涂在尼龙网上并蒸发溶剂的方式得到厚度约60 μm的复合电解质膜,室温离子电导率为2.4×10-4S/cm。Liu 等[69]通过将LPSC 溶于有机碱溶液中,在颗粒上涂覆了5 nm 厚的均质聚多巴胺层,通过冷压得到厚度为35 μm的自支撑LPSC 薄膜,其室温离子电导率为0.2 mS/cm。还有的研究者通过添加Li盐的方式当做聚合物增塑剂,但往往也难以将离子电导率突破10-4S/cm[70-71]。虽然与聚合物混合之后的硫化物SSEs 在柔性、厚度、电化学窗口、电池稳定性等方面有所优化,但无一例外地降低了离子电导率。
作为聚合物黏结剂的一个大类,橡胶(rubber)因其较高的黏度和在非极性溶剂中的良好溶解性,已被大量使用于硫化物SSEs 电解质膜的制备中,主要包括:丁苯橡胶(styrene butadiene rubber,SBR)、丁腈橡胶(NBR)等。Lee 等[72]早在2017 年就分别测试了不同黏结剂与非极性溶剂对75Li2S-25P2S5电解质成膜的影响,并对比了干法与湿法合成两种不同路线。最终使用5%的NBR与邻二甲苯作为黏结剂和溶剂的电解质表现出几乎无损的离子电导率和优秀的电池性能。而NBR 具有较强黏性的原因是其中的—C≡N 官能团与硫化物的Li+发生离子-偶极相互作用,可以让硫化物在复合膜中良好分散。随后该团队基于一种嵌段共聚物(styrenebutadiene-block-copolymers,SBS)橡胶,通过“点击反应”(click reaction)引入羧基(—COOH)合成了一种新型SBS黏结剂(SBS-COOH)[73]。将仅有2%的SBS-COOH添加入复合正极或LPSC电解质中,电极的黏附性未受到影响,离子电导率降低幅度较小(约2.6 mS/cm)。全电池循环后电极和电解质内部出现裂痕颗粒数量大量减少[图5(a)]。而Emley 等[74]基 于DLVO 理 论(derjaguin-landauverwey-overbeek theory)[图5(b)],探究了分散体的固体负载与浇铸成型的LPSC薄膜质量之间的相关性。在LPSC∶NBR的比例固定为20∶1时,使用50∶50(质量比)的甲苯和异丁酸异丁酯作为溶剂,当LPSC在溶剂中的比例为54%时,最后制得厚度约50 μm 的电解质膜具有约1.12 mS/cm 的几乎无损的室温离子电导率和十分均匀的颗粒分散。Zhu 等[75]将LPSC 与1%的硅橡胶(silicone rubber,SB)溶于氯仿(chloroform)后球磨,将混合后的浆料刮涂在纤维素骨架上并在干燥后冷压得到厚度约60 μm的电解质膜。纤维素骨架提高了电解质膜的机械强度和柔韧性,表现出对硫化物颗粒良好的附着性,并且可以大面积制备。所得到的复合电解质室温离子电导率可达6.3 mS/cm,在S||Li-In 固态电池中比冷压的Pellet-type电解质表现出更好的性能[图5(c)]。除橡胶之外,纤维素(cellulose)及其衍生物也可用作黏结剂。Cao 等[76]使用2%的乙基纤维素与LPSC 制备了厚度仅约47 μm 的电解质膜。乙基纤维素独特的两亲分子结构既可以溶于甲苯,又能保持与LPSC 的稳定性。复合电解质膜在30 ℃下的离子电导率为1.65 mS/cm,与970 μm厚的LPSC pellet 离子电导率相当(1.67 mS/cm),但极低的阻抗值(3.43 Ωvs.60.37 Ω)表现出sheettype电解质的巨大优势[图5(d)]。
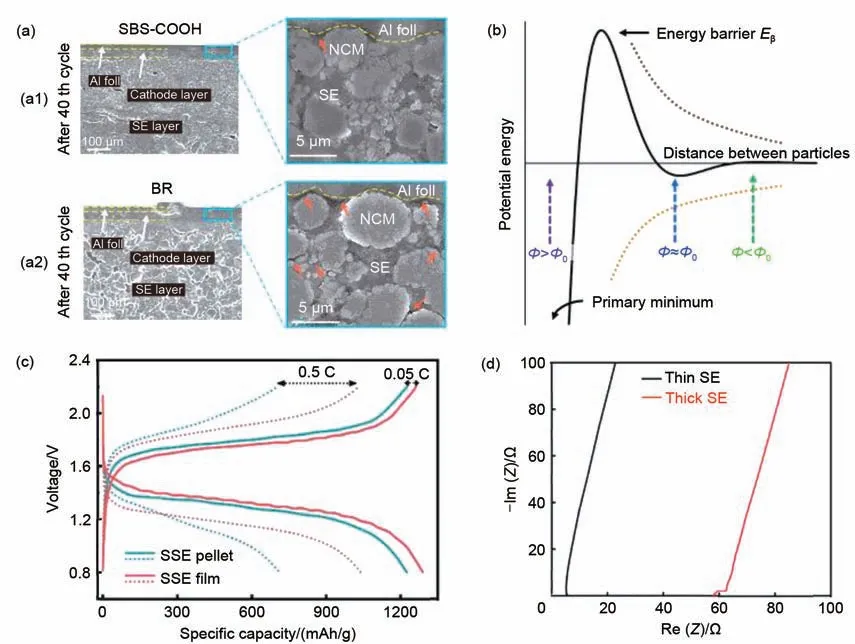
图5 (a)使用(a1)SBS-COOH和(a2)BR黏结剂的电池在第40个循环后的SEM截面图[73];(b)由DLVO理论解释的分散粒子的势能和存在两个能量极小值作为分散粒子之间距离的函数[74];(c)分别使用SSE膜和SSE颗粒的S||Li-In电池在0.05和0.5 C时的恒流充放电曲线[75];(d)薄的和厚的SSEs在交流阻抗测试中的Nyquist图[76]Fig.5 (a)Cross-sectional SEM images of the batteries based on(a1)SBS-COOH and(a2)BR binders after the 40th cycle[73];(b)Potential energy of dispersed particles and the existence of two energy minima as a function of distance between particles in a dispersion as explained by the DLVO theory[74];(c)Galvanostatic chargedischarge profiles at 0.05 and 0.5 C of S//Li-In cells with the SSE film and the SSE pellet[75];(d)Nyquist plots in AC impedance measurement of the thin and thick SSEs[76]
除了作为黏结剂之外,聚合物还可以只作为骨架(scaffold)来提升硫化物SSEs 的柔韧性。Nam等[77]将聚对苯二甲酰胺(PPTA)无纺布用作骨架材料,冷压在甲苯溶液成型的Li3PS4膜两侧,形成厚度约70 μm的NW-LPS-NW三明治结构。尽管电导率值略有下降(1.6×10-4S/cm),但具有较低的界面阻抗和柔韧性。PPTA 骨架的高柔韧性确保了复合材料的机械完整性,并防止了由于结构缺陷或孔隙造成的断裂。Xu 等[78]将Li3PS4的甲苯溶液滴加在Kevlar 无纺布上制备了约100 μm 厚的电解质膜,室温离子电导率为3×10-4S/cm。类似的,Kim等[79]将Li6PS5Cl0.5Br0.5的乙醇溶液渗透入静电纺丝的聚酰亚胺(PI)骨架中,约70 μm 的电解质膜可以达到2×10-4S/cm的离子电导率。
2.2 干法制备的复合电解质
相比于湿法制备,干法制备的复合电解质虽然减少了溶剂的加入,避免了潜在的有毒溶剂使用或溶剂对硫化物SSEs结构的破坏,但增加了混合均匀的难度,容易导致聚合物黏结剂发生团聚或阻碍硫化物SSEs 本征的离子传输[72]。如何高效利用干法来制备复合电解质成为了需要更加深入探索的问题。
在早期阶段,由于玻璃型硫化物SSEs 的离子电导率通常不高,所以有的研究集中在了提升离子电导率的方向上并取得了一定成效。Hayashi等[80-81]尝试了多种低聚物与Li7P3S11电解质通过高能球磨混合来制备复合电解质。实验证明,仅添加量2%(物质的量分数)的1,4-丁二醇就可以提高复合电解质相对于纯电解质的离子电导率(9.7×10-5S/cm)。1,4-丁二醇的加入不仅可以与玻璃型LPS电解质之间形成P—O—C 键,还能够降低玻璃化转变温度(Tg),这些是离子电导率提升的关键原因。而使用链段更短的乙二醇可以实现更高的室温离子电导率(1.1×10-4S/cm)。对于聚合物含量较多的复合电解质而言,状况通常是顾此失彼。Whiteley等[82]使用原位交联工艺,在77.5Li2S-22.5P2S5电解质中添加20%的不同聚亚胺粉末,通过热压成型固态电解质膜,可以达到约63 μm的厚度,室温离子电导率为9.2×10-5S/cm。Villaluenga 等[83]使用Li3PS4玻璃电解质和23%的全氟聚醚聚合物(PFPE)制备了厚度150~290 μm 的电解质膜,室温离子电导率约10-4S/cm,离子迁移数接近1。由于一些聚合物具有良好的分散性和连通性,填充了硫化物SSEs的空隙并大大提高了复合电解质的延展性和结构稳定性。然而,过量地添加聚合物使得这些协同设计还是在一定程度上牺牲了硫化物SSEs的离子电导率。
早在2003年,Inada等[84]就将0.01Li3PO4-0.63 Li2S-0.36SiS2和Li3.25Ge0.25P0.75S4与硅橡胶(SR)、丁苯橡胶(SBR)分别通过湿法与干法制备电解质。作者发现通过干法制备的SBR 复合电解质比湿法制备的表现出更高的电导率。湿法制备的复合电解质颗粒上的SBR 会阻断Li+的传导。而通过干法制备可以实现SBR 颗粒在电解质中的均匀分散,实现了电解质颗粒之间的良好接触。由于可以纤维化的性质,聚四氟乙烯(PTFE)黏结剂的使用在近年来逐渐成为热点[21]。Hippauf等[85]将LPSC与0.15%的PTFE混合并在研钵中手磨30 min以上,通过热压方法得到电解质膜。Zhang等[86]使用Li5.4PS4.4Cl1.6电解质与0.2%的PTFE 通过球磨法混合,再通过对辊机获得厚度30 μm的电解质膜,室温离子电导率达到了8.4 mS/cm[图6(a)]。以NCM523为CAM组装的全电池在60 ℃和0.05 C的倍率下循环150圈后的放电比容量为108.5 mAh/g,容量保持率80.2%。Jiang 等[87]将质量比为100∶1 的LGPS 与PTFE 混合并辊压在尼龙网上,获得了厚度约100 μm 的电解质膜,获得了3.6×10-4S/cm的室温离子电导率。Wang 等[88]使用PTFE 作为黏结剂,实现了LPSC、Li3InCl6、Li6.5La3Zr1.5Ta0.5O12(LLZTO)等不同种类的固态电解质膜的大面积制备(8 cm×6 cm),厚度低至15~20 μm[图6(b)]。基于Li3InCl6和LPSC 的双层电解质膜组装Li3InCl6@LCO/Li3InCl6+LPSC/Graphite@LPSC 的全固态软包电池的初始放电比容量为121.2 mAh/g(0.1 C),50次循环后容量保持在83.1 mAh/g。能够大面积制备sheet-type电解质和电极的无溶剂方法可能是一种可行的技术,可以促进固态电池的生产从实验室研究过渡到工厂制造,并进一步向商业化迈进。

图6 (a)超薄SSE膜制备示意图[86];(b)20 μm厚的LLZTO、Li3InCl6和Li6PS5Cl电解质膜的照片[88]Fig.6 (a)Schematic illustration of the ultrathin SSE membrane fabrication[86];(b)Photo of LLZTO,Li3InCl6 and Li6PS5Cl membranes with thickness of 20 μm[88]
3 全固态电池中的硫化物复合正极
在全固态电池中,正极通常由正极活性材料(CAMs)、固体电解质和导电添加剂三种成分组成,电化学反应通常在固-固界面进行,差的固-固接触会产生较大的界面阻抗[51]。有别于准固态电池的解决方法,研究者们还提出了其他的策略来实现硫化物SSEs 与正极的良好配合,并且往往会借助有机溶剂来溶解(dissolve)或分散(disperse)硫化物SSEs,或使用有机黏结剂来制备Sheet-type 电极。
3.1 通过溶液法改善复合正极内部界面
溶液法通常需要将硫化物SSEs 溶解于溶液之中,或者通过原位液相法合成硫化物SSEs,在去除有机溶剂后,会更容易在CAMs和硫化物SSEs前驱体溶液之间形成一层均匀的界面。Tatsumisago课题组的研究人员[89-90]基于N-甲基甲酰胺(NMF)液相合成了Li3PS4电解质、基于乙醇合成了LPSC电解质,并分别使用NMF和乙醇溶液将LPS和LPSC电解质涂覆在LiCoO2颗粒上,形成了电极界面的良好接触。同样,LPSC电解质还可以用乙醇加乙腈的双溶液进行溶解,并通过混合液相球磨的方法涂敷在NCM111颗粒上,乙腈作为一种表面活性剂可以实现纳米级颗粒的沉淀[91]。然而正极颗粒上的这种直接涂层并不能抑制界面副反应,电池容量和倍率性能的提高主要是由于CAMs的利用率提高和复合正极中电荷转移动力学的增强。Park等[23]使用均相甲醇溶液制备了一种空气稳定的玻璃型电解质0.4LiI-0.6Li4SnS4,并涂敷在LiNbO3@LiCoO2颗粒上,与固体混合LPS电解质的复合正极相比,溶液混合的电池表现出更小的极化,并且在暴露于空气后,充放电曲线只产生了极其微小的变化。Wang 等[92]通过两步法在LiCoO2颗粒上构建了一种双壳保护层:首先使用原子层沉积(atomic layer deposition,ALD)将LiNbO3包覆在LiCoO2上,再将分散于正庚烷的LGPS 包覆在LiNbO3@LiCoO2上[图7(a)]。其中内壳LiNbO3抑制界面反应,而外壳LGPS 实现紧密的电极-电解质接触。因此,双壳结构正极具有125.8 mAh/g(0.1 C)的初始比容量和90.4%的首圈库仑效率。Han 等[93]在乙醇中溶解Li2S、聚乙烯吡咯烷酮(PVP)和LPSC 电解质,然后通过共沉淀和高温碳化过程制备出了一种纳米正极复合材料[图7(b)]。均匀分散的纳米级正极颗粒具有较好的力学性能和离子/电子导电性,所组装的全电池在循环60 圈后仍具有830 mAh/g(50 mA/g)的可逆比容量。
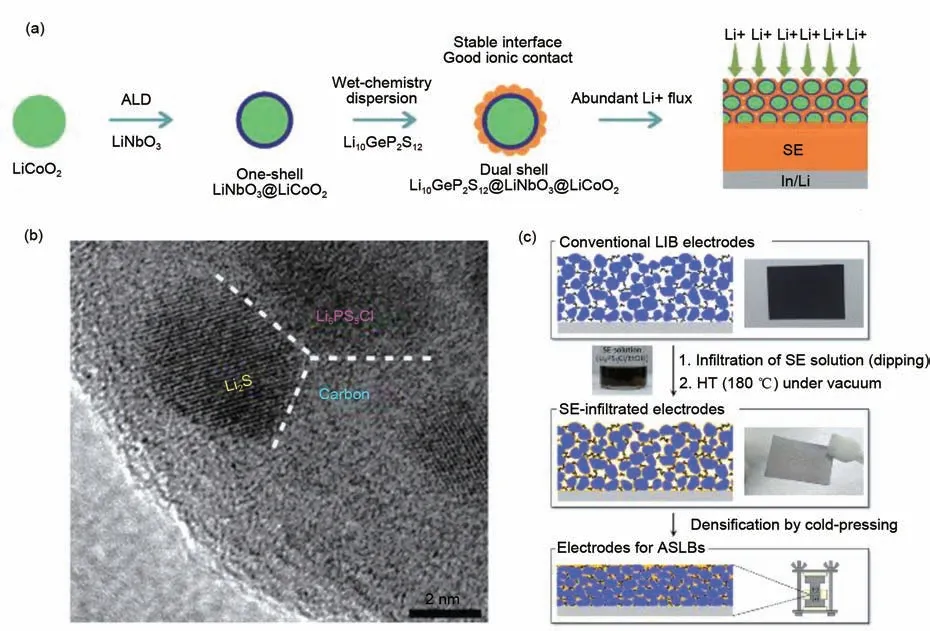
图7 (a)双壳的LGPS@LNO@LCO合成示意图[92];(b)制备的Li2S-LPSC-C 纳米复合材料的高分辨TEM照片[93];(c)常规锂离子电池复合电极的溶液渗透工艺示意图[98]Fig.7 (a)Schematic diagram of synthesis of dual shell LGPS@LNO@LCO[92];(b)The high-resolution TEM image of the as-obtained Li2S-LPSC-C nanocomposite[93];(c)Schematic diagram of the infiltration process of conventional LIB composite electrodes[98]
玻璃型硫化物SSEs 通常可被用于原位合成。Lin 等[94]使用纳米Li2S 与P2S5在THF 中通过表面反应制备了含有Li3PS4超离子导电涂层的NanoLi2S。减小的CAM 粒径和超离子导电涂层改善了正极材料的离子电导率和内部阻抗。Yao 等[95]通过将Co9S8、Li2S与P2S5溶解在乙腈中再热处理的方式,将原位合成的纳米级Li7P3S11锚定在Co9S8纳米片上。组装的固态电池在1000圈后仍拥有421 mAh/g(1.27 mA/cm2)的比容量。通过同样的方法还可以合成Fe3S4@Li7P3S11、MoS2@Li7P3S11等硫化物纳米复合正极[96-97]。除了通过溶液方法得到复合正极粉末再组装Pellet型全固态电池,研究者们逐渐开始从片状集流体电极片入手,实现复合正极的制备。Kim 等[98]将溶于乙醇的LPSC 浸涂在传统锂离子电池电极上,实现了电极内部紧密的颗粒接触,再通过冷压方法使之致密化之后,实现了6%~8%的低孔隙率[图7(c)]。这种基于传统电极片的渗透法体现了在“卷对卷”制造工艺中的适用性。
3.2 Sheet-type复合正极的制备
由于对固态电池质量能量密度和体积能量密度越来越高的要求,科研人员不得不逐渐减小复合正极和固态电解质的厚度[88]。探究制备sheet-type 的复合正极的工艺也成为了研究的热点之一。
在sheet-type电极制作的初期阶段,大多像传统的锂离子电池正极刮涂制备工艺一样,将混合有硫化物SSEs的正极浆料刮涂在铝箔为主的集流体上。作为一种性能较好的弱极性有机黏结剂,NBR在正极侧也获得了较多应用。Jung 课题组就在NBR 的使用上做了很多探索。先后曾使用THF、二甲苯、二溴甲烷以及二溴甲烷与丁酸丁酯的配合溶剂,通过浆料混合的方式将NCM与硫化物SSEs复合的正极刮涂在铝箔上,所制备的电极片可用于组装软包电池(pouch cell)[39,99-101]。对于NBR 而言,用溶液法制备电极不仅能获得更薄的电极,还在于比干法电极拥有更好的分散性和离子接触[72]。除NBR 之外,一种共聚物SBS 橡胶也有部分应用[73,102]。Zhang 等[103]还报道了使用1%的乙基纤维素作为黏结剂来刮涂NCM811 和LPSC 的复合正极,其中用到了乙醇作为溶剂。为了克服黏结剂分子极性与硫化物SSEs 不兼容的问题,Lee 等[104]使用聚(丙烯酸叔丁酯)-b-聚(1,4-丁二烯)(TBA-b-BA)作为黏结剂,设计了一种原位“脱保护”方法:首先将叔丁基基团保护的非极性TBA-b-BA 与NCM711、LPSC、super P分解在丁酸丁酯中刮涂电极片,通过热处理的方式裂解叔丁基以获得—COOH,黏结剂具有极性之后可以形成氢键,将NCM颗粒与铝箔的黏合性大大提升[图8(a)]。有别于使用铝箔集流体的负载方式,基于PTFE黏结剂的能够自支撑(free-standing)的复合正极也可以通过黏结剂和干法制造工艺的改善被生产出来[85,88]。通过加入≤5%的PTFE并且通过研钵手动研磨即可获得大面积的复合正极和负极膜。而作为对电池能量密度的极大优化,拥有双极堆叠(bipolar stacking)的固态电池也可以通过制作sheet-type的电极和电解质而被组装出来。Cao 等[105]使用1.5%和4%的乙基纤维素作为黏结剂,分别通过抽滤的方式制备了NCM 复合正极和Si 复合负极膜,并基于LPSC 电解质膜组装了双极堆叠全固态电池[图8(b)]。组装的双极全固态电池可以在4.5~8.2 V 范围内充放电,并获得了145 mAh/g(0.1 C)的首次放电比容量和70.7%的首圈库仑效率[图8(c)]。双极固态电池的组装无疑对规模制备的Pouch电池组装有着积极的意义。通过双极堆叠将单电池单元串联在全电池内,可以减少电芯中的集流体、包装、导线等材料的使用,不但大大提高电池的能量密度和功率密度,也同时降低了电池生产的成本[14,106-107]。
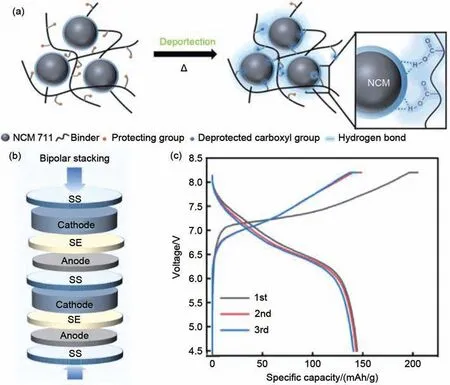
图8 (a)复合正极中聚合物黏结剂和活性材料之间的保护-脱保护化学原理图[104];(b)双极堆叠电池的组装示意图;(c)双极电池前三圈的充/放电曲线[105]Fig.8 (a)Schematic illustration of the protection-deprotection chemistry between the polymeric binder and active material in the composite cathode[104];(b)The fabrication of bipolar stacked battery through layer-by-layer stacking;(c)Charge/discharge profiles of bipolar battery at the first three cycles[105]
4 结 语
硫化物固态电解质作为下一代高比能电池最有潜力的固态电解质之一,有着巨大的优势,但是其较差的化学稳定性限制了批量制备和大规模电池组装。综合全文所述,有机物对硫化物SSEs的增强作用主要体现在:
(1)改善的电极/电解质界面接触和相应阻抗的降低;
(2)增强了硫化物SSEs 的柔韧性,拓宽了电压窗口,并促进了部分硫化物的离子电导率;
(3)使硫化物SSEs 更好独立或配合CAMs 成膜,以供大面积制备。
对于准固态电池和基于湿法制备的复合正极或复合电解质来说,最大的困难出现在溶剂的选择上,现有的电池工业所用大部分溶剂(NMP、DMF、THF 等)和电解液(DMC、DME 等)都具有较强极性,用于溶解或分散硫化物SSEs后通常会造成难以挽回的离子电导率损失,并且也难以溶解弱极性的聚合物黏结剂。对于添加电解液来说,虽然离子液体和溶剂化离子液体已经被证明对硫化物相对稳定,但吡咯等离子液体的使用和需要基于摩尔比大量添加的Li 盐会进一步增加电池制造的成本,并且一些易燃溶剂的使用会增大安全隐患。对聚合物黏结剂来说,传统的强极性聚合物(PVDF、PEO等)无法为硫化物SSEs提供足够的黏性,且需要极性溶剂溶解;而弱极性的黏结剂(NBR、纤维素等)则因为需要较多的添加,容易造成对SSEs 或CAMs的表面覆盖,从而阻止了离子/电子迁移。故而需要找到易于硫化物电解质膜制备的弱极性黏结剂,其在辅助成膜的同时应尽量减小对SSEs离子电导率的影响。作者认为未来的有机物辅助增强硫化物基固态电池的研究应主要集中在复合正极、电解质以及两者界面处,并可能呈以下趋势:
(1)探究使用微量、具有特定成膜等功能性且对硫化物SSEs稳定的电解液,以改善电极/电解质界面和复合正极内部界面;
(2)探究使用微量、易分散且黏性大的聚合物黏结剂,以制备柔性的高性能电极或电解质膜。
猜你喜欢
上海理工大学学报(2021年3期)2021-07-20 08:04:04
陶瓷学报(2021年1期)2021-04-13 01:33:40
陶瓷学报(2021年1期)2021-04-13 01:32:54
山东冶金(2019年5期)2019-11-16 09:09:12
电源技术(2016年2期)2016-02-27 09:04:59
中国资源综合利用(2016年7期)2016-02-03 03:00:19
中国资源综合利用(2016年7期)2016-02-03 03:00:11
环境科技(2015年3期)2015-11-08 12:08:36
电源技术(2015年9期)2015-06-05 09:36:06
导航定位学报(2015年2期)2015-06-05 09:27:42
