
限域型贵金属氧还原反应电催化剂研究进展
2022-07-07 02:12:36胡冶州王得丽
储能科学与技术 2022年4期
胡冶州,王 双,申 涛,朱 叶,王得丽
(1香港理工大学应用物理系,香港 999077;2能量转换与存储材料化学教育部重点实验室,华中科技大学化学与化工学院,湖北武汉 430074)
质子交换膜燃料电池具有环境友好、能量密度高和能量转换效率高等优点,被认为是极具发展前景的一类能量转换装置[1-4]。然而缓慢的阴极氧还原反应(ORR)动力学极大程度上限制了质子交换膜燃料电池的规模化应用[5-6]。因此,开发高效的阴极氧还原催化剂具有重要意义。贵金属催化剂,如Pt/Pd 基催化剂,仍为目前性能最优、应用最为广泛的氧还原反应电催化剂,然而考虑到其有限的储量以及昂贵的价格,实现其良好催化活性下的服役寿命延长至关重要。为实现这一目标,近年来的研究主要集中在以下几个方面:①晶面调控[7-8],通过选择性暴露特定晶面实现性能最优;②电子结构调控[9-10],通过引入第二种元素进行合金化以实现对Pt/Pd电子结构的调控;③形貌调控[10-13],一方面可以减少贵金属用量,另一方面可以通过构筑具有特定维度的形貌以实现最大化暴露活性位点。虽然上述策略均能够在一定程度上改善催化剂活性及稳定性,但是在催化剂使用过程中,催化剂与强酸性电解质的直接接触必然会导致催化剂的溶解腐蚀,这一现象在贵金属合金催化剂体系中尤为明显[14]。另外由于金属溶解导致的颗粒脱落、团聚等问题同样难以忽视[15]。根据美国能源部2020 年技术指标,铂族金属催化剂稳定性应满足如下条件:经过30000 圈循环后,0.9 V 电位下的质量比活性衰减率小于40%。根据目前的文献来看,仍然有较大部分催化剂难以实现这一指标。基于此,本文综述了限域型贵金属氧还原反应电催化剂的设计策略,通过将贵金属催化剂“固定”起来,不仅抑制了催化剂在强酸性环境中的溶解,同时限制颗粒在载体上的迁移及脱落,最大程度保留了活性位点,从而提升催化剂稳定性。
1 限域型贵金属氧还原反应催化剂设计
近年来,限域型ORR 催化剂在酸性电解质中展现出明显提升的ORR 稳定性。这是因为与未限域的催化剂相比,限域型催化剂能够有效抑制电催化过程中的团聚及脱落等问题,而颗粒的团聚及脱落问题是造成催化剂衰减的主要原因。另外,限域层很大程度上避免了催化剂与强酸电解质之间的直接接触,这使得催化剂腐蚀速率减慢。尤其是对于Pt基合金催化剂而言,其中的过渡金属通常具有更负的标准电极电势,相对于Pt更容易发生溶解。因此在限域型贵金属催化剂中,限域层能够缓解过渡金属溶出问题,避免了电化学循环过程中过渡金属快速溶出导致的催化剂稳定性衰减问题。研究发现,限域层的选择对催化剂电化学行为产生不同程度的影响,并且制备策略的差异也会产生具有不同构型的限域型催化剂(图1)。目前,限域层的选择主要分为以下四种类型;①导电聚合物;②非金属氧化物;③金属氧化物;④碳层。因此,本文总结了不同限域层包覆的贵金属ORR 催化剂的研究进展,介绍了不同限域层对催化剂电化学行为的影响,着重分析了不同限域策略对催化剂稳定性的提升机制。
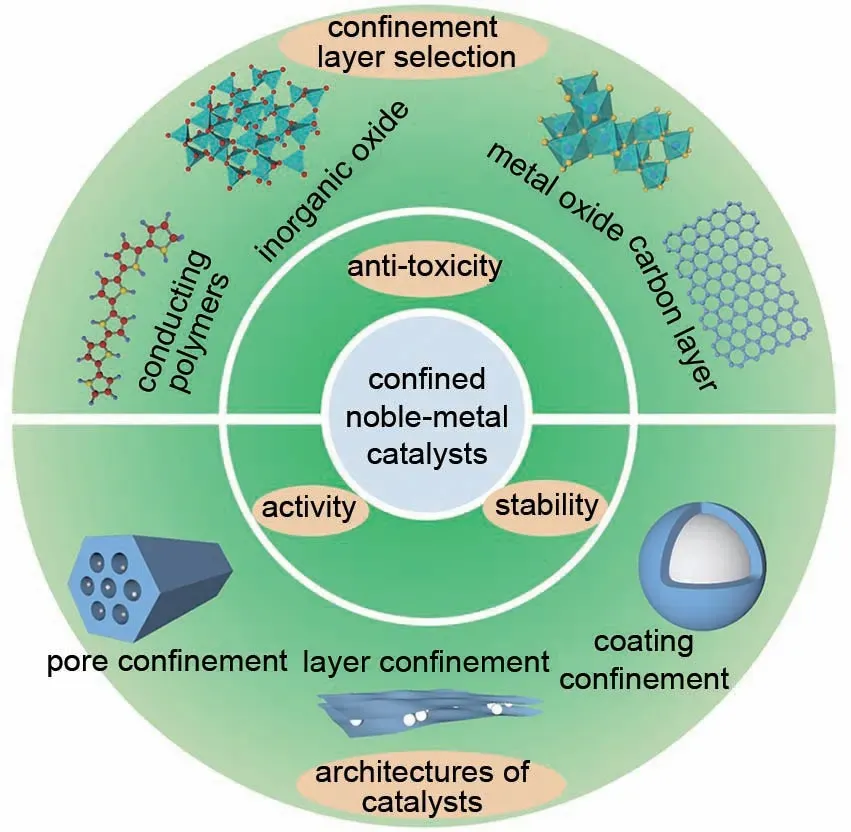
图1 限域层的选择及限域催化剂的构型Fig.1 The selections of confined layers,and architectures of confined noble-metal catalysts
1.1 导电聚合物限域
导电聚合物,如聚吡咯、聚苯胺等,由于其具有独特的化学稳定性以及优异的导电性,被广泛用于传感器、光电以及催化等领域[16]。此类限域催化剂的制备通常在室温下即可进行,在一定引发剂存在的情况下,聚合物单体即可完成聚合,并在目标催化剂表面形成限域层。Zhang等[17]以V2O5纳米纤维凝胶作为模板及氧化剂,实现吡咯聚合的同时,完成Pt离子的还原及限域,得到了一种具有夹层结构的铂纳米颗粒-聚吡咯纳米纤维催化剂(Pt NP@PPy NF)[图2(a)]。聚吡咯除了作为导电网格实现Pt与反应物之间的电子传递外,聚吡咯包覆层的限域作用还有效抑制了Pt纳米颗粒在酸性电解质溶液中的溶解和脱落。Mizuhata等[18]以苯胺作为包覆层前驱体,进而得到聚苯胺限域的Pt纳米颗粒催化剂(Pt/PPy/C)。在0.5 mol/L H2SO4中,循环500圈前后的ORR 峰电流几乎不变,但是未限域的Pt/C催化剂在500 圈循环后ORR 峰电流几乎消失。尽管结果表明聚苯胺的包覆有效抑制了颗粒在电催化过程中的团聚。但是,由于难以控制聚苯胺包覆层厚度,使得Pt纳米颗粒与反应物之间的接触受限,大量的Pt纳米颗粒不能充分利用。
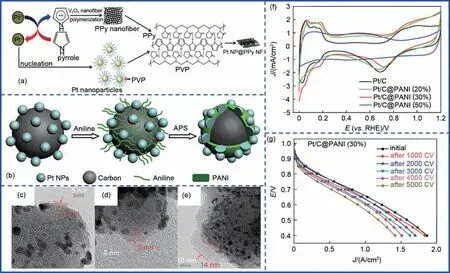
图2 导电聚合物限域。(a)Pt NP@PPy NF合成示意图;(b)Pt/C@PANI合成示意图;(c)~(e)不同厚度聚苯胺包覆层的Pt/C@PANI的TEM图及(f)CV曲线对比;(g)Pt/C@PANI催化剂在不同循环圈数下的燃料电池极化曲线对比Fig.2 Conducting polymers confinement.(a)Schematic diagram of Pt NP@PPy NF;(b)Schematic diagram of Pt/C@PANI;(c)—(e)TEM images and(f)CV curves of Pt/C@PANI with different thickness of coating layer;(g)MEA stability test of Pt/C@PANI(30%)
为改善这一问题,Wei等[19]将Pt/C分散至一定浓度的苯胺单体的硫酸溶液中,利用苯胺单体与碳载体之间的π-π共轭作用选择性地将苯胺单体优先吸附在碳载体上,然后在(NH4)2S2O8(APS)的氧化作用下,实现苯胺单体在碳载体上的原位聚合,进而得到具有核壳结构的聚苯胺包覆的Pt纳米颗粒催化剂(Pt/C@PANI)[图2(b)],并且通过调控苯胺前驱体量得到了不同厚度聚苯胺包覆层[图2(c)~(e)]。结果表明,当苯胺载量控制在30%时,包覆层厚度约为5 nm,催化剂能够展现出最优异的ORR性能,随着包覆层厚度的增加,CV 曲线中的氢特征峰完全消失[图2(f)]。这是因为在5 nm聚苯胺包覆层情况下,Pt/C与含氧物种的结合强度能够得到有效的调控,Pt的d轨道与聚苯胺包覆层的π共轭配体促进了二者之间的电子转移,使得该催化剂的抗氧化能力得到显著提升。而在酸性溶液中稳定存在的聚苯胺包覆层则很大程度上避免了碳载体与酸性环境的直接接触,因此有效抑制了由于碳载体腐蚀而导致的Pt 催化剂的脱落等问题。相较于Pt/C 催化剂,Pt/C@PANI 催化剂在质子交换膜燃料电池中展现出明显提升的循环稳定性,0.6 V 电位下,经过5000圈循环,电流密度仅衰减了24%[图2(g)]。
1.2 非金属氧化物限域
非金属氧化物因具有较好的热稳定性和耐腐蚀性而被作为包覆层应用于限域催化剂的制备中,进而提升催化剂稳定性。然而非金属氧化物通常较为致密,这不利于反应传质的进行,将表面非金属氧化物多孔化则能够有效避免这一问题。
2007 年,Takenaka 等[20]首次报道了一种多孔SiO2限域的Pt/CNT 催化剂(SiO2/Pt/CNT)的制备方法,利用表面底剂3-氨丙基三乙氧基硅烷(APTES)以及四乙基硅烷(TEOS)在CNT 上的连续水解,通过后续高温热处理成功实现了SiO2在Pt/CNT 上的包覆[图3(a)]。电化学CV 曲线上可以看出,SiO2/Pt/CNT展现出与Pt/CNT相似的氢吸、脱附峰,且循环1000圈后,CV曲线没有发生明显变化,表明包覆的SiO2层能够在不影响Pt 的电化学行为的前提下,提升催化剂的稳定性[图3(b)]。对比SiO2/Pt/CNT 与Pt/CNT 催化剂循环过程中ECSA 的变化可以看出,循环1300 圈后,SiO2/Pt/CNT 催化剂的ECSA 仅从44 m2/g 衰减至35 m2/g[图3(c)]。这主要是SiO2层的包覆有效抑制了催化剂使用过程中颗粒的溶解、迁移以及团聚等问题。尽管如此,部分致密的SiO2层仍然阻隔了部分活性位点,使得在ORR活性上相较于Pt/CNT仍有一定程度差距,这是因为由TEOS衍生的SiO2包覆层通常包括四面体的SiO4结构单元,其中两个硅原子共同连接一个氧原子从而组成类似沸石结构的孔结构,不利于O2的传输。Takenaka 等[21]发现将Si 源由TEOS 替换为甲基三乙基氧基硅烷(MTEOS)则能够很好地改善这一问题[图3(d)~(e)],这是因为在TEOS 中乙氧基易水解产生Si―O―Si环状结构,而在MTEOS中由于相互作用较强的Si―C键的存在而不易水解。因此,MTEOS衍生的SiO2包覆层具有较大的孔径分布。此外,由于烷基基团的存在,此类SiO2包覆层比TEOS衍生的SiO2层具有更强的憎水性,有利于ORR过程中水从催化剂表面脱去,进而促进ORR进行。电化学测试也表明MTEOS衍生的SiO2包覆的Pt/CNT[SiO2/Pt/CNT(MTEOS)]相较于TEOS 衍生的SiO2包覆的Pt/CNT[SiO2/Pt/CNT(TEOS)],不仅具有更为优异的ORR活性,同时ORR稳定性也得到提升[图3(f)]。这种通过改变硅前驱体种类来改善SiO2层孔径分布以及亲水性的策略被认为能够有效提高活性位点的暴露数量并促进最终产物在催化剂表面的脱附过程。Aoki等[22]发现,在SiO2包覆过程中,将表面底剂APTES 替换为巯基丙基三乙氧基硅烷(MPTES)能够有效减少SiO2在碳载体上的沉积含量,并进一步抑制纳米颗粒团聚长大,这是因为与APTES 中的-NH 相比,MPTES 中的-SH与Pd、Pt 等金属之间具有更强的相互作用。电化学测试及TEM 表征表明,具有较低SiO2含量的SH-SiO2/Pt/Pd/C 催化剂在限制电催化过程中颗粒的团聚方面上体现出一定优势。
刻蚀后的蒙脱石(e-MMT)也被作为限域层抑制Pt 纳米颗粒的迁移及团聚,提升催化剂稳定性。Wei 等[23]利用e-MMT 与Pt(NH3)22+之间的离子交换作用实现了Pt离子在MMT插层间的原位还原,得到了MMT 限域的Pt 纳米颗粒(Pt/e-MMT)[图3(g)]。1600圈循环后,Pt/e-MMT-40%的ECSA仅衰减了31%,而Pt/C的ECSA则衰减了49%[图3(h)、(i)]。循环后的TEM图也表明Pt/e-MMT中的Pt纳米颗粒平均粒径几乎没有发生明显变化。除了限域作用外,MMT 与Pt 纳米颗粒之间的相互作用也在一定程度上抑制了Pt纳米颗粒的溶解及脱附,从而使得该催化剂即便在强酸条件下仍具有优异的化学稳定性。
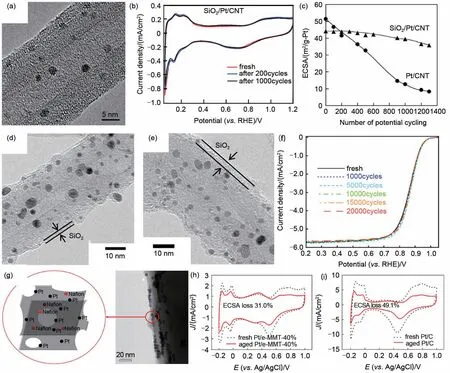
图3 非金属氧化物限域。(a)以TEOS为硅源得到的多孔SiO2限域的Pt/CNT催化剂(SiO2/Pt/CNT);不同循环圈数下SiO2/Pt/CNT的(b)CV曲线对比及(c)ECSA变化曲线;(d)~(e)以MTEOS为硅源得到的SiO2限域的Pt/CNT催化剂[SiO2/Pt/CNT(MTEOS)];(f)SiO2/Pt/CNT(MTEOS)循环不同圈数后的LSV曲线;(g)MMT限域的Pt纳米颗粒(Pt/e-MMT)示意图;(h)Pt/e-MMT和(i)Pt/C催化剂循环前后的CV曲线对比及ECSA大小变化Fig.3 Inorganic oxide confinement.(a)TEM image of SiO2/Pt/CNT using TEOS as precursor;(b)CV curves and(c)ECSA change of SiO2/Pt/CNT at different potential cycles,(d)—(e)TEM images of SiO2/Pt/CNT(MTEOS)using MTEOS as precursor;(f)ORR stability test of SiO2/Pt/CNT(MTEOS);(g)TEM image of Pt/e-MMT;The CV curves of(h)Pt/e-MMT and(i)Pt/C before and after stability tests
1.3 金属氧化物限域
与非金属氧化物相比,金属氧化物与Pt之间的较强的金属-载体相互作用能够有效提升Pt的催化性能[24]。而通过金属氧化物在Pt表面的包覆则可以很好地将金属-载体相互作用以及包覆策略优势进行结合,进一步提升催化性能。Cheng等[25]采用原子层沉积(ALD)方法并通过不同次数的ALD循环实现了不同厚度ZrO2包覆的Pt 纳米颗粒(ALDxZrO2-Pt/NCNT,x为循环次数)[图4(a)]。其中ALD50ZrO2-Pt/NCNT 经过600 ℃处理后,Pt 纳米颗粒平均粒径仅从1.8 nm 变为2.0 nm,而ALD20ZrO2-Pt/NCNT中Pt纳米颗粒粒径则从1.8 nm变为4.2 nm,说明合适厚度的ZrO2限域能有效抑制Pt 纳米颗粒高温下的团聚。不仅如此,ZrO2的包覆在提升催化剂电催化循环稳定性上也发挥了显著作用,经过4000 圈的循环稳定性测试发现ALD50ZrO2-Pt/NCNT 催化剂ECSA 相较于初始状态仅衰减8%,而Pt/NCNT 则衰减了74%[图4(b)]。TiO2凭借其优异的化学稳定性及与Pt之间强的金属-载体相互作用得到广泛研究。Lu等[26]报道了一种多孔TiO2限域的Pt纳米颗粒催化剂(Pt-TiO2)[图4(c)],在0.5 mol/L H2SO4溶液中循环3000 圈后,Pt-TiO2催化剂的极限电流密度几乎没有发生衰减。然而金属氧化物普遍存在导电性差以及在酸性溶液中不稳定的问题,在电催化过程中往往难以发挥催化作用,不利于ORR 反应中传质的进行,因此在电催化反应过程中的“去金属氧化物包覆层”就显得至关重要。Sun 等[27]先合成了单分散的fcc-FePt,随后在其表面包覆MgO 得到了fcc-FePt/MgO,对其进一步高温热处理及酸刻蚀得到了fct-FePt纳米颗粒。MgO限域层有效抑制了fcc-FePt合金高温有序化过程颗粒的团聚问题[图4(d)~(e)]。通过对fcc-FePt 在硫酸溶液中进行Fe溶出测试[图4(f)],可以看出MgO先包覆后刻蚀得到的fct-FePt纳米颗粒中Fe溶出得到明显抑制。

图4 金属氧化物限域。(a)ZrO2限域的Pt纳米颗粒制备示意图;(b)ALDPt/NCNT、ALD50ZrO2-Pt/NCNT600 ℃和E-TEK Pt/C三种催化剂在不同循环圈数下的ECSA变化曲线;(c)经过加速循环稳定性测试后的ALD50ZrO2-Pt/NCNT的STEM图;(d)MgO包覆的fcc-FePt纳米颗粒;(e)去除MgO包覆层后的fct-FePt纳米颗粒;(f)0.5 mol/L H2SO4溶液中,fcc-FePt与fct-FePt催化剂中Fe含量随时间变化曲线;(g)~(h)氧化铁包覆的Pd2FeCo/C催化剂低倍下的STEM图;(i)Pd2FeCo/C与Pd/C催化剂循环前后的ORR性能对比Fig.4 Metal oxides confinement.(a)Schematic diagram of ZrO2 confined Pt nanoparticles;(b)The normalized ECSA of ALDPt/NCNT,ALD50ZrO2-Pt/NCNT600 ℃and E-TEK Pt/CPMTT at different cycling stages;(c)The STEM image of ALD50ZrO2-Pt/NCNT after stability test;(d)MgO confined fcc-FePt nanoparticles;(e)fct-FePt nanoparticles after removal of MgO;(f)The Fe composition in fcc-FePt/C and fct-FePt/C at different times;(g)—(h)STEM images of Fe2O3 confined Pd2FeCo nanoparticles;(i)The stability test of Pd2FeCo/C and Pd/C
尽管这种利用金属氧化物层的限域作用抑制颗粒团聚长大的策略具有较好的效果,但目前金属氧化物包覆的制备方法较为复杂且一般需要经过后续去除步骤,因此很大程度上限制了此类催化剂的规模化应用。Wang 等[28]通过简单的浸渍-还原法在Pd2FeCo/C纳米颗粒热退火过程中自发形成了仅为1.1 nm 厚度的Fe2O3包覆层[图4(g)]。在该包覆层的限域作用下,经过退火过程后的Pd2FeCo纳米颗粒粒径大小仅为6.5 nm[图4(h)],有效避免了由于颗粒团聚等所造成的活性位点减少等问题。由于Fe2O3易溶于酸性溶液中,因此在电化学测试过程即可将其去除,无需额外的后处理过程,简化了催化剂制备流程。电化学测试中可以看出,由于自发形成的Fe2O3层对Pd2FeCo催化剂的包覆以及合金化对Pd的应力调控,该催化剂ORR活性、稳定性均具有一定程度的提升[图4(i)]。然而随着反应的进行,Fe2O3包覆层逐渐溶解,必然会导致催化剂与强酸电解质溶液间的直接接触,这会加速合金的溶解腐蚀,造成催化剂性能进一步衰减。
1.4 碳层限域
近年来,碳层限域型贵金属催化剂得到了研究人员的广泛关注,碳层限域一方面能够有效抑制纳米颗粒在合成及电催化过程中的团聚长大等问题,另一方面,得益于其丰富的孔隙结构,在实际使用中,合理的催化剂设计无需经过繁琐的去除步骤,这极大地简化了催化剂的设计成本。目前碳层限域贵金属催化剂的设计主要有以下三种思路:①“沉积-转化”策略,即在贵金属催化剂表面先沉积上合适厚度的碳层前驱体,并通过后续高温热处理得到碳层限域的贵金属催化剂;②“嵌入-转化”策略,即先设计出具有丰富孔隙结构的高比表面积碳,其不仅作为导电载体,同时作为物理限域层,并通过后续贵金属前驱体在高温下的还原和扩散作用自发形成碳纳米孔限域的贵金属催化剂;③“一步热解法”,即一锅法热处理碳限域层与贵金属混合的前驱体直接得到碳层限域的贵金属催化剂。
1.4.1 “沉积-转化”策略
“沉积-转化”策略实现碳层在催化剂表面的包覆是一个多步骤过程,通常基于以下思路进行:首先是贵金属催化剂的常规合成,通过水热反应或聚合反应完成碳层前驱体在催化剂表面的吸附,最后通过高温碳化得到碳层限域的贵金属催化剂。
Li 等[29]以葡萄糖作为碳层前驱体及还原剂,采用水热法将Pt沉积在SBA-15孔道内,通过后续高温碳化以及模板去除,得到了介孔碳限域的Pt纳米颗粒(Pt@C/MC)[图5(a)]。由于介孔碳层的限域作用有效抑制了Pt纳米颗粒的团聚、脱落等问题,因此在经过长时间的稳定性测试后,该催化剂的氧还原峰电流仅衰减了4%。
聚苯胺由于其丰富的N含量,将其高温碳化后可直接得到氮掺杂石墨化碳。Wei 等[30]以聚苯胺作为氮源以及碳源,利用其在Pt/C催化剂表面原位聚合得到聚苯胺包覆的Pt/C,通过后续在惰性气氛下900 ℃高温碳化得到氮掺杂石墨化碳包覆的Pt/C(Pt/C@NGC)[图5(b)~(c)]。其中原位产生的多孔氮掺杂碳层不仅能够有效地抑制高温过程中Pt纳米颗粒的迁移、团聚,还能够在电催化过程中抑制Pt纳米颗粒的脱落、溶解等问题。除此以外,氮掺杂石墨化碳层还能够作为第二活性位点,并通过对Pt电子结构的调控提高ORR 活性。所制备的Pt/C@NGC在酸性溶液中经过加速循环稳定性测试后仍然保持较高催化性能,且相比于初始状态,电化学活性面积仅衰减了约8%,远小于Pt/C催化剂电化学活性面积的衰减(41%)。通过循环前后的TEM表征发现,Pt/C@NGC 催化剂循环后粒径仅发生微弱变化,从平均粒径4.7 nm变为5.4 nm,而Pt/C催化剂的平均粒径由2.8 nm变为7.8 nm。
多巴胺具有一定的黏附性,几乎可以附着在所有物质表面,在弱碱性条件下,通过自身氧化聚合可得到聚多巴胺[31]。Hyeon 等[32]利用多巴胺在无序PtFe 合金上的包裹,通过后续高温处理,得到氮掺杂碳层包覆的PtFe 有序金属间化合物[图5(d)]。原位产生的氮掺杂碳层能够能有效防止PtFe 合金在高温热处理过程中的团聚,将颗粒大小限制在6.5 nm 左右。并且通过调控多巴胺在PtFe 合金上的聚合时间,可以精确地控制碳层的厚度,发现当碳层厚度控制在1 nm 以下时,氮掺杂碳层可以在保护PtFe 合金的同时保持较高的渗透性,从而使反应物与催化剂表面的接触不受影响[图5(e)]。所得到的氮掺杂碳包覆的有序PtFe 合金(fct-PtFe/C)催化剂无论是在氧气饱和还是氩气饱和气氛下,经过加速循环稳定性测试后,该催化剂均展现出优异的ORR稳定性。当进行全电池测试时,fct-PtFe/C催化剂在连续工作100 h后的最大功率密度仅衰减了3.4%,与之相比,Pt/C催化剂则衰减了27%[图5(f)]。通过原位XANES,EDS以及DFT计算发现,氮掺杂碳层和有序PtFe合金结构对于fct-PtFe/C催化剂稳定性的提升至关重要。Sun等[33]先通过微波辅助多元醇法制备了PtNi3/C 催化剂,并在其表面包覆一定厚度的聚多巴胺层,通过后续高温热处理,得到了氮掺杂碳层限域的PtNi3纳米颗粒(PtNi3/C@NGC)。电化学性能测试表明,PtNi3/C@NGC催化剂在0.1 mol/L HClO4溶液中展现出优异的ORR活性以及循环稳定性。
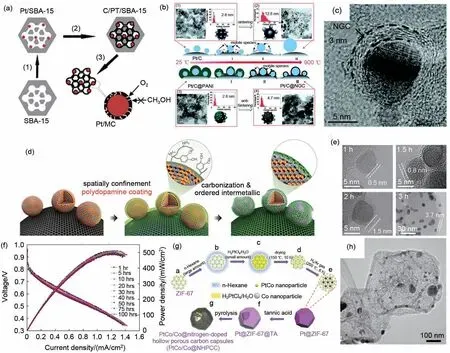
图5 基于“沉积-转化”策略的碳层限域。(a)Pt@C/MC的制备示意图;(b)Pt/C@NGC的制备示意图;(c)单个Pt/C@NGC的HRTEM图;(d)碳包覆L10-PtFe合金的制备示意图;(e)不同碳层厚度包覆的L10-PtFe纳米颗粒的TEM图;(f)碳包覆L10-PtFe合金的稳定性测试;(g)PtCo/Co@NHPCC的制备示意图;(h)PtCo/Co@NHPCC的TEM图Fig.5 Carbon confinement base on‘deposition-conversion’strategy.(a)Schematic diagram of Pt@C/MC;(b)Schematic diagram of Pt/C@NGC;(c)The TEM image of Pt/C@NGC;(d)Schematic diagram of L10-PtFe nanoparticles;(e)The TEM images of L10-PtFe nanoparticles with different thickness of carbon layers;(f)The stability test of carbon confined L10-PtFe nanoparticles in MEA;(g)Schematic diagram of PtCo/Co@NHPCC;(h)The TEM image of PtCo/Co@NHPCC
单宁酸作为一类来源广泛的植物多酚,其价格低廉,在中性及微碱性条件下,可附着大多数物质表面形成包覆层。Chen 等[34]将具有一定亲水性的ZIF-67 作为载体吸附Pt 前驱体,通过氢气还原先得到ZIF-67负载的Pt纳米颗粒(Pt@ZIF-67),随后以单宁酸作为包覆层前驱体沉积在Pt@ZIF 表面,通过后续热处理即可得到具有中空结构的氮掺杂碳包覆的PtCo 合金(PtCo/Co@NHPCC) [图5(g)~(h)]。其中外层包覆的单宁酸碳化后所产生的中空多孔碳结构不仅有效保持了ZIF 模板的初始形貌,同时还促进了ORR过程中电子转移以及传质过程,因此相较于Pt/C 催化剂,PtCo/Co@NHPCC 展现出更为优异的ORR活性和稳定性。
1.4.2 “嵌入-转化”策略
通过对碳载体孔隙结构的精准设计,利用介孔碳中丰富的孔隙结构所提供的限域作用同样能够实现对Pt纳米颗粒的包覆。Ryoo等[35]以SBA-15作为硬模板,糠醇作为碳源,通过简单的浸润法得到的孔道被糠醇聚合物所填充的SBA-15 前驱体。并通过后续高温碳化及模板的去除,得到了一种有序介孔碳纳米阵列[图6(a)]。将其作为限域载体负载Pt催化剂,不仅实现了Pt纳米颗粒的均匀分散,同时该介孔碳的限域作用使得Pt纳米颗粒具有更小的粒径,因此相较商业Pt/C,该催化剂展现更为优异的ORR性能。
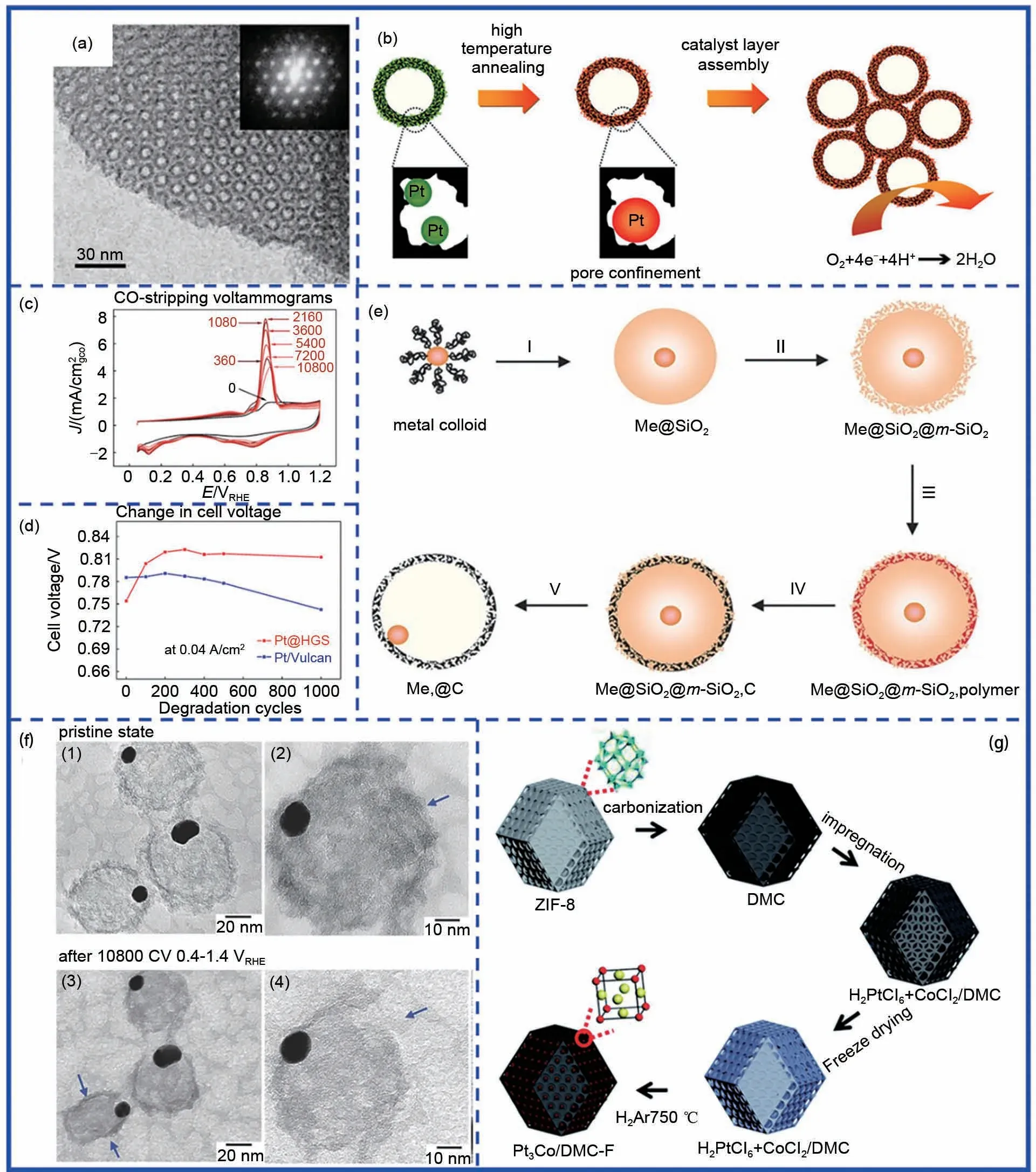
图6 基于“嵌入-转化”策略的碳层限域。(a)一种有序介孔碳纳米阵列的TEM图;(b)Pt@GHS的制备示意图;(c)不同循环圈数下Pt@GHS的CO剥离曲线;(d)Pt@GHS催化剂循环过程中燃料电池电压变化曲线;(e)AuPt@C催化剂的制备示意图;(f)AuPt@C催化剂循环前后的TEM图;(g)Pt3Co/DMF-F的制备流程示意图Fig.6 Carbon confinement base on‘insertion-conversion’strategy.(a)The TEM image of ordered nanoporous arrays of carbon;(b)The schematic diagram of Pt@HGS;(c)The CO stripping curves of Pt@HSG at different potential cycling;(d)Comparison of cell voltage change of Pt@HGS and Pt/Vulcan during stability test;(e)Schematic diagram of encapsulated metal nanoparticles in porous carbon shells;(f)IL-TEM images of AuPt@C before and after stability test;(g)Schematic diagram of Pt3Co/DMF-F
Galeano 等[36]报道了一种中空介孔石墨化碳包覆的Pt 纳米颗粒的制备方法。以二氧化硅作为模板,在Ar 气饱和溶液中将硝酸铁、二乙烯基苯(DVB)以及偶氮二异丁腈(AIBN)嵌入二氧化硅介孔中实现DVB 的聚合,通过后续高温碳化以及模板去除得到具有高比表面积(>1000 m2/g)、高度石墨化以及孔径分布集中的交联介孔结构的中空石墨化碳球(HGS),最后通过浸渍法将Pt前驱体沉积到制备的HGS 孔中,经过高温煅烧热处理得到颗粒大小均匀介孔碳包覆的Pt纳米颗粒(Pt@GHS)[图6(b)]。由于所制备的HGS 具有超高比表面积,使得高温煅烧后的Pt@GHS中Pt纳米颗粒能够实现在HGS上的均匀分布,且3~4 nm的孔径有效地抑制了高温热处理过程中颗粒长大。经过长时间的加速循环稳定性测试后,Pt@GHS的ECSA衰减程度明显小于Pt/Vulcan[图6(c)]。全电池测试发现,在1000圈的启动-终止过程中,Pt@GHS的电池电压几乎没有发生变化,而Pt/C 则衰减了约50 mV [图6(d)]。通过对Pt@HGS 进行原位扫描电镜表征以及相同位置透射电镜表征,发现经过3600 圈循环后,除了部分负载于表面的颗粒有所减少外,位于介孔内部的Pt颗粒密度以及粒径几乎没有发生变化,进一步说明中空石墨化碳球对Pt的包覆作用能够有效限制催化剂团聚以及脱落的问题。2014年,Galeano等[37]对该方法进行拓展后制备了一种具有核壳结构的碳层包覆AuPt 合金(AuPt@C)[图6(e)],并测试了其在较高电压范围内的循环稳定性。通过相同位置透射电镜表征发现,由于碳层包覆的限域作用,循环前后AuPt 颗粒大小几乎没有发生明显变化,仅有碳层发生微小的变化[图6(f)]。
Zhao等[38]以ZIF-8热解得到的介孔碳作为限域载体,并通过后续浸渍、冻干以及高温还原得到介孔碳限域的Pt3Co 金属间化合物(Pt3Co/DMF-F)[图6(g)]。这种ZIF-8热解得到的介孔碳展现出高达1619 m2/g 的比表面积以及较窄的孔径分布,这有利于克服高温有序化过程颗粒的团聚长大以及粒径分布不均等问题,同时氮掺杂碳与金属之间的相互作用也有效避免了催化剂在电催化过程中脱落而导致的性能衰减问题。这种将催化剂嵌入介孔碳的策略为制备粒径较小的金属间化合物提供了新思路。然而这种基于“嵌入-转换”的包覆策略由于制备过程较为复杂,难以通过一步法实现,这导致大量能耗产生,不利于规模化生产。
1.4.3 “一步热解”策略
“一步热解”策略能够在一次加热处理过程中同时完成碳前驱体碳化及贵金属的还原,步骤相对简单,能耗也能够极大程度减少,有利于进一步降低此类催化剂的生产成本。通过简单调控反应物前驱体量可轻易实现不同厚度碳层限域的贵金属催化剂,这有利于针对不同贵金属催化剂实现最优碳层厚度的包覆。Zhao 等[39]利用化学沉积法实现了一步Pt还原、碳层沉积以及高温石墨化的过程,通过后续模板去除得到有序介孔碳包覆的Pt 纳米颗粒(Pt@GC)[图7(a)]。相较于CMK-3负载的Pt纳米颗粒而言,Pt@GC催化剂展现出更为优异的ORR活性和稳定性。然而此类硬模板的去除步骤必然会导致催化剂制备过程复杂化。
Lee等[40]利用有机配体与Pd离子之间的配位作用,先通过1-萘胺与Pd配位形成层状配位聚合物,然后将得到的配位聚合物直接碳化得到具有框架结构的碳包覆Pd 催化剂(Pd@N-C NFs)[图7(b)]。热解过程中,层状Pd-NA 前驱体转变为多孔框架结构,Pd 以约5.5 nm 粒径均匀分布在碳骨架上且被较薄的碳层所包覆。其中1-萘胺作为氮源和碳源,不仅有利于氮掺杂碳的生成,更有利于金属催化剂与氮掺杂碳基底之间实现更紧密的接触。其次金属离子与含氮配体之间的相互作用也有利于防止热处理过程中颗粒的团聚、长大等问题。萘胺中杂原子掺杂有利于产生多功能杂原子掺杂碳,进一步提升电催化性能。Wan 等[41]以(NH4)2S2O8为引发剂,PANI为碳源与H2PtCl6和CNT在低温条件下进行聚合,并通过后续高温碳化、酸洗得到氮掺杂多孔碳限域的Pt 纳米颗粒(Pt@CNx/CNT) [ 图7(c)]。Pt@CNx/CNT 在酸性条件下循环1500 圈以后,半波电位几乎没有发生明显衰减。这主要是由于氮掺杂碳层对Pt纳米颗粒的限域作用避免了其在载体上的脱落、团聚等。类似地,Kwon等[42]将Pt-苯胺配位化合物与CNF 进行均匀混合,通过后续一步高温热处理得到CNF 负载的碳层限域Pt 纳米颗粒(Pt@CS/CNF)。通过改变碳化温度对碳层石墨化程度进行调控发现,900 ℃处理后的Pt@CS/CNF展现出最优的ORR稳定性[图7(d)],意味着除了对碳层厚度进行合理调控外,还应关注其石墨化结构。Wang 等[43]以双氰胺作为碳层前驱体,与H2PtCl6、FeCl2及Vulcan碳黑均匀混合后,通过一步高温热处理得到了超薄氮掺杂碳限域的PtFe 有序合金(O-PtFe@NC/C)[图7(e)]。一步高温热处理中双氰胺先在较低温下转变为石墨化C3N4,随着温度的进一步升高,C3N4进行分解,并完成在PtFe合金表面的原位碳层包覆,同时无序PtFe 进一步转变为有序PtFe 合金。结果表明,碳层限域一方面抑制了合金高温有序化过程中的颗粒团聚问题,还能够有效抑制毒化物种如SOx、POx等在催化剂表面的吸附,从而提高催化剂的抗毒化性能,在160 ℃下,O-Pt-Fe@NC/C组装的高温质子交换膜燃料电池的最大功率密度高达384 mW/cm2[图7(f)]。利用类似的方法,Wang 等[6]实现了多孔氮掺杂碳限域的有序Pd-Fe合金催化剂(O-Pd-Fe@NC/C)。考虑到Pd 相较于Pt具有相对较低的氧化还原电位(Pd: 0.951 V,Pt: 1.18 V),我们发现碳限域层需要达到相对较厚的状态才能够一定程度上提高Pd基催化剂的ORR稳定性。得益于碳层限域作用,所得O-Pd-Fe@NC/C 催化剂的MA 衰减程度明显降低,且抗CO及SOx性能也得到明显改善。
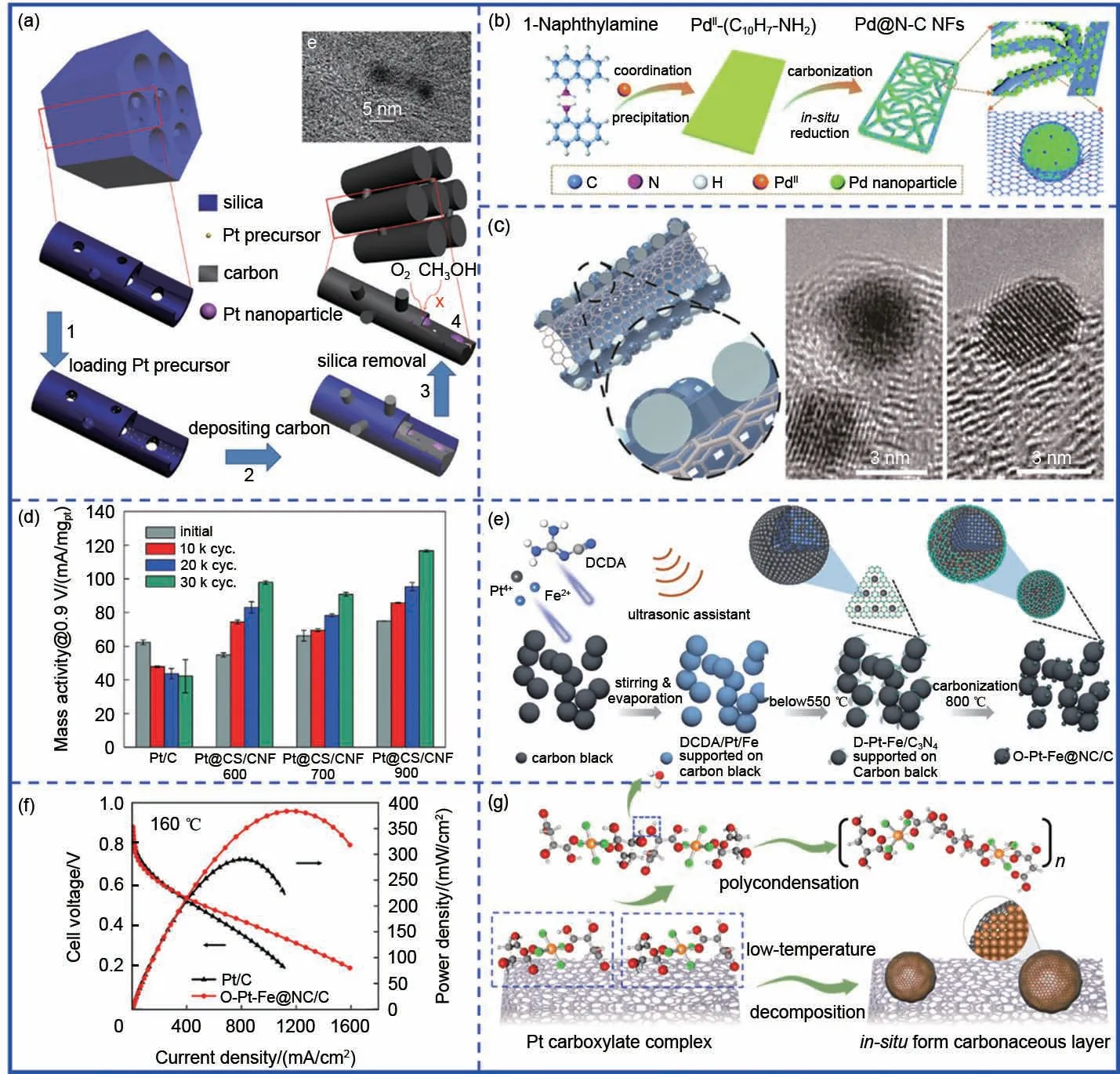
图7 “基于一步热解策略”的碳层限域。(a)Pt@GC的制备示意图;(b)Pd@N-C NFs制备示意图;(c)Pt@CNx/CNT的制备示意图;(d)Pt@CS/CNF的稳定性测试对比图;(e)O-PtFe@NC/C的制备示意图;(f)O-PtFe@NC/C催化剂的高温质子交换膜燃料电池极化曲线;(g)一步低温热处理得到Pt@C/C的制备示意图Fig.7 Carbon confinement base on‘one-step pyrolysis’strategy.(a)The schematic diagram of Pt@GC;(b)The schematic diagram of Pd@N-C NFs;(c)The schematic diagram of Pt@CNx/CNT;(d)The stability tests of Pt@CS/CNF;(e)The schematic diagram of O-Pt-Fe@NC/C;(f)The high-temperature fuel cell polarization curves of O-Pt-Fe@NC/C and Pt/C;(g)The synthesis of Pt@C/C trough one-step low temperature pyrolysis
目前用于实现碳层限域的碳前驱体普遍需要高温热处理,进而实现其在催化剂表面的包覆。高温一方面意味着更高的能耗,另一方面不可避免导致部分未成功包覆的颗粒发生团聚。针对这一问题,Wang等[44]以苹果酸作为碳前驱体,将其与H2PtCl6和Vulcan 碳均匀混合后,于150 ℃真空热处理实现了低温下碳层在贵金属的表面包覆(Pt@C/C)[图7(g)]。电化学结果表明,碳层限域不仅有效提高催化剂的ORR 稳定性,还减弱了催化剂与毒化小分子之间的吸附强度,从而具有更优的抗毒化性能。循环前后的XPS 测试发现,Pt 氧化态含量几乎没有明显变化,这意味着碳层限域还能够有效抑制金属的氧化所造成的活性位点减少。进一步地,合成了40%高载量的Pt@C/C催化剂,并将其应用于高温质子交换膜燃料电池中。在160 ℃条件下,该电池表现出662 mW/cm2的峰值功率密度。
2 结论和展望
综上所述,对于贵金属ORR 催化剂而言,通过将其限域在物理保护层内的策略能够有效抑制催化剂的团聚、脱落、溶解及氧化等所导致的稳定性衰减问题。本文综述了不同包覆层限域的贵金属催化剂,包括导电聚合物限域、非金属氧化物限域、金属氧化物限域及碳层限域(图8)。对于导电聚合物限域的贵金属催化剂而言,此类催化剂的制备通常能够在较低温度下进行,根本上避免了高温造成的催化剂颗粒团聚等问题,并且能够显著降低催化剂制备过程的能耗,有利于规模化制备。其难点在于如何合理控制包覆层在贵金属催化剂上的均匀包覆,从而实现贵金属的高效利用。同时提升导电聚合物与催化剂之间的黏附力也至关重要,这能够有效延长限域层的作用时间,进一步提升催化剂稳定性。对于非金属氧化物限域的贵金属催化剂而言,非金属氧化物限域在酸性电解质中具有极佳的抗腐蚀能力,然而非金属氧化物通常不具有良好的导电性,不利于此类催化剂在电催化ORR 中的应用。金属氧化物限域的贵金属催化剂除了能够通过限域作用抑制催化剂的团聚等问题外,金属氧化物与贵金属之间的强相互作用还能够对贵金属催化剂的电子结构进行调控,避免催化脱落导致的稳定性衰减问题。然而金属氧化物的包覆过程通常较为复杂,且因金属氧化层导电性较差及易腐蚀,对于此类催化剂,金属限域层的作用更多的是抑制催化剂在高温过程中的团聚。除此以外,此类限域型催化剂的循环圈数测试普遍较少,难以准确评判其长时间的循环稳定性。因此后续研究应更加准确地评估其长时间稳定性。对氧还原这一三相界面电催化反应而言,传质性及导电性至关重要。尽管导电聚合物限域、非金属氧化物限域及金属氧化物限域能够有效抑制颗粒团聚及脱落所造成的稳定性衰减问题,这些限域层的致密性、绝缘性很大程度上限制了这些催化剂使用场景。与其他限域策略相比,碳层限域具有如下优势:①碳层前驱体来源广泛,有利于设计出不同性质碳层限域的贵金属催化剂,从而实现对催化剂性能的优化;②碳材料本身具有优异的导电性,在碳层限域的贵金属催化剂中往往能够加快电子的传递,促进反应的进行;③碳限域层本身可以作为活性位点,对碳层进行杂原子掺杂能够轻松调控限域层的催化性能,从而有利于与贵金属催化剂产生协同作用促进ORR 进行。但是目前碳层限域催化剂的制备主要是在高温下进行,且碳层包覆厚度难以实现精准控制。此类催化剂的循环稳定性及活性仍有待进一步优化,因此未来的研究重点应放在以下方面:①实现对表面包覆层厚度的精准控制,优化包覆层孔结构,提供限域作用的同时保证传质的进行;②优化限域型贵金属催化剂制备策略,将制备过程简易化,这有利于催化剂规模化制备。
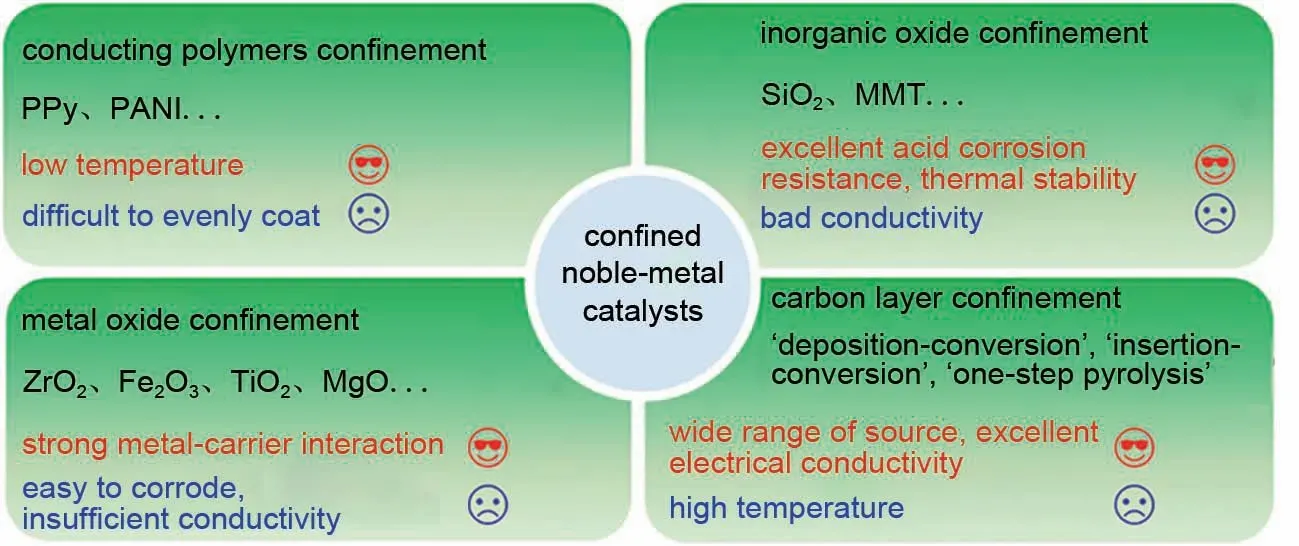
图8 限域型贵金属催化剂应用于电催化氧还原反应Fig.8 Confined noble-metal catalysts for efficient ORR
猜你喜欢
证券市场周刊(2024年13期)2024-04-16 04:33:35
贵金属(2021年1期)2021-07-26 00:39:20
贵金属(2021年1期)2021-07-26 00:39:20
潍坊学院学报(2021年2期)2021-07-22 07:59:04
——庆祝中国共产党成立一百周年贵金属纪念币展
中国钱币(2021年4期)2021-02-26 00:58:18
山西化工(2019年4期)2019-02-17 09:36:46
物理学报(2019年1期)2019-01-25 09:53:52
物理化学学报(2018年1期)2018-01-29 07:45:45
载人航天(2016年3期)2016-06-04 06:08:42
浙江工商大学学报(2015年3期)2016-01-07 06:51:07
