
转录组和蛋白质组整合分析揭示组蛋白H2A去泛素化酶USP16的功能调控网络
2022-07-05 03:44张旭敏
复旦学报(自然科学版) 2022年3期
华 杰, 吴 真, 张旭敏
(复旦大学 生命科学学院,上海 200438)
在真核生物中,基因组DNA与组蛋白和非组蛋白结合在一起组成染色体的基础结构单元核小体。核小体由147 bp的基因组DNA和组蛋白八聚体组成,其中包括两份组蛋白H2A,H2B,H3和H4[1]。相邻核小体由组蛋白H1附着的DNA连接起来,形成串珠状结构,进而形成更高级的结构。染色质在结构上高度动态,以便进行复制、转录、重组、DNA损伤修复和其他以DNA为基础的生物学过程。在调控染色质动态性和可及性的多种机制中,组蛋白的翻译后修饰扮演了重要的角色。组蛋白修饰包括甲基化、磷酸化、乙酰化、泛素化和许多新发现的组蛋白修饰如SUMO化等[2-3]。这些修饰调节了相邻核小体之间的相互作用,同一染色体中核小体之间的作用,不同染色体中核小体之间的相互作用,对于所有与染色质相关的生理活动都有重要的影响,进而影响了一系列的生理活动,如细胞增殖、细胞分化、细胞凋亡等。
泛素化是将单个或者多个泛素(ubiquitin)组成的链连接到蛋白质上的过程,分别被称为单泛素化和多泛素化。泛素化主要发生于赖氨酸和蛋白质N端的甲硫氨酸上,偶尔也会发生于半胱氨酸,苏氨酸和丝氨酸上[4]。泛素化由连续的酶活反应催化: 由泛素激活酶E1连接泛素,E1将泛素转移到泛素结合酶E2上,泛素连接酶E3将泛素从E2转移到特定的底物上,泛素连接酶E3决定了泛素化底物的特异性[4]。去泛素化是将泛素从蛋白质上去除的过程,这一过程由特异的去泛素化酶催化。人类细胞中已经发现了100多种去泛素化酶,它们通过复杂的调节机制参与不同的生物学过程中。人类基因组中还有许多基因拥有与已经鉴定的去泛素化酶同源的序列,亦可能作为去泛素化酶发挥功能[5]。去泛素化酶在细胞周期调控、细胞膜信号转导、基因转录、DNA损伤修复等过程中发挥了重要的作用[5-7]。去泛素化酶的调控一般涉及到蛋白复合物组装、酶活性调节、亚细胞定位等过程[8-9]。
在所有组蛋白上都已经发现了泛素化的存在[2]。在这其中,组蛋白H2A的泛素化(H2Aub)是最重要的,细胞内大约有10%~15%的组蛋白H2A会发生泛素化。尽管在组蛋白H2A的K129和K15上都发现了泛素化的存在,但泛素化主要发生于K119位置(H2AK119ub)[10-14]。这一修饰在许多重要的生理过程中都起到作用,比如转录调控、DNA损伤修复、异染色质形成、X染色体失活等[10-12,15-18]。
组蛋白H2A作为第一个被发现的泛素化蛋白,其泛素化和去泛素化已经被研究了相当长的时间,组蛋白H2AK119位置的泛素化和去泛素化对于多种生命活动至关重要。之前的研究发现H2AK119ub主要由多梳抑制复合物1(PolycombRepressiveComplex 1, PRC1)催化,而其去泛素化主要由USP16(Ubiquitin carboxyl-terminalSpecificPeptidase 16)催化[13,15,19]。许多由H2AK119ub调控的基因对于细胞分化、细胞分裂等过程至关重要,研究H2AK119ub调控的基因表达能够增进我们对相关调节机制的了解,以及为相关疾病的治疗提供理论策略。
USP16是属于USP家族并在动物中保守的半胱氨酸蛋白酶,包含一个Zn-F结构域和两个UBP结构域,通过C204位点的半胱氨酸攻击连接泛素与蛋白质赖氨酸位点的共价键发挥去泛素化酶功能[20]。USP16在1999年由Cai等鉴定为一种新的去泛素化酶,他们发现这种去泛素化酶与有丝分裂密切相关,故命名为Ubp-M(Ubiquitin-processing protease-Mitosis),该去泛素化酶的野生型定位于细胞之中,但C204S活性缺失突变体定位于细胞核中[20]。有趣的是,C204S突变体的过表达会抑制细胞的有丝分裂,使细胞逐渐停止分裂并发生凋亡,而过表达野生型并不会出现这样的抑制效应[20]。当时人们猜测,USP16也许是一种在有丝分裂期间运动到细胞核内发挥去泛素化酶功能的蛋白质,但是其底物和调节的信号通路并不为人们所知。直到2007年,Joo等[21]通过体外去泛素化体系寻找组蛋白H2A去泛素化酶,他们从HeLa细胞的核提取物中鉴定到了能对H2AK119ub发挥去泛素化酶功能的成分,最后分离到了Ubp-M并将其更名为USP16,并证明USP16是核小体中组蛋白H2A特异的去泛素化酶,USP16通过去泛素化H2AK119ub调控细胞周期和基因表达[22]。后续有关于USP16的研究几乎都集中于USP16在细胞核中通过去泛素化H2AK119ub发挥功能,调控如干细胞的分化、细胞分裂、细胞周期等生物学过程[13,19,21]。USP16过表达能够抑制干细胞自我更新,促进细胞衰老,这种调控可能与唐氏综合征的发病机制有关[23]。usp16在白血病发生过程中,是致癌转录因子RUNX1的靶基因,可能与造血系统发挥功能有关[24]。usp16敲除的小鼠在胚胎期死亡,且骨髓中的条件敲除表明它对造血干细胞发挥功能是必需的[19]。USP16在多项研究中都被证明调控了DNA损伤修复过程,但有人认为USP16的蛋白剂量变化会影响DNA损伤修复[25],也有人认为USP16的蛋白剂量变化与DNA损伤修复无关[26]。
尽管USP16在病理学中的重要作用日益明确,但USP16被证明在细胞周期的所有阶段都主要存在于细胞质中,USP16被细胞积极地排除在细胞核外,在G2/M期特定进入细胞核中发挥功能[22]。USP16对H2K119ub的去泛素化过程是许多组蛋白修饰的前置条件,如H3S10磷酸化等[21]。出人意料的是,USP16没有典型的核定位信号,某些研究表明USP16可能在细胞质中也发挥了重要的功能,例如USP16可能通过调节PLK1的稳定性促进线粒体-微管连接的形成[27-28]。
鉴于USP16主要定位于细胞质中,但在细胞核内发挥功能,迄今对于USP16下游的功能网络和信号途径缺乏深入研究。我们认为有必要对USP16下游的调控网络和信号途径从组学层面进行整体性的分析,因此我们利用在HeLa细胞中表达靶向usp16的shRNA抑制其蛋白表达,并利用嘌呤霉素筛选,Western-blot鉴定得到将USP16的蛋白表达量明显降低的稳定细胞株,然后通过组学整合分析探究USP16调控的下游网络。通过将转录组学和蛋白质组学数据进行联合分析,我们对USP16调控的基因在组学层面有了整体性的认识,明确了USP16特异调控的细胞内生物学过程,揭示了USP16调控的下游基因网络,发现了USP16在调控细胞周期和线粒体功能中潜在的重要功能,为进一步深入研究提供了理论依据。
1 材料与方法
1.1 材料
HeLa和293T细胞由美国阿拉巴马大学伯明翰分校药学院王恒彬副教授提供。usp16敲低的细胞系通过向HeLa细胞转染表达shRNA的质粒后通过嘌呤霉素(puromycin)筛选得到,该实验在王恒彬副教授实验室完成。载有靶向usp16的shRNA载体(pHTP-shRNA-USP16),表达带有RFP(Red Fluorescent Protein)标签的USP16的载体(pcDNA3-RFP-USP16)由本实验室保存。抗USP16抗体由本实验室保存,anti-GAPDH抗体(Ab8245)购自Abcam。本文转录组和蛋白质组数据来源于两株经筛选后得到的usp16敲低细胞株。
1.2 方法
1.2.1 细胞培养
本文中涉 添加了10 %胎牛血清(Gibco 10099)和200 U / mL青霉素-链霉素(Gibco 15140)的DMEM培养基(Gibco 8119130),筛选出的KD-1和KD-3细胞需要在完全培养基中添加2 μg / mL嘌呤霉素。所有细胞均在含有5 %CO2的37℃细胞培养箱中培养。
1.2.2usp16敲低细胞株的构建
本实验室已经构建了靶向usp16的shRNA载体pHTP-shRNA-USP16,其编码shRNA的DNA序列[21]为5′-ACCAGTGCTTAGAGAACTATCTCTTGAATAGTTCTCTAAGCACTGGT-3′(下划线标记的为靶向USP16的序列)。
将pHTP-shRNA-USP16质粒在E.coliDH5α菌种中扩增培养后纯化质粒。待10 cm培养皿中细胞密度为70%~90%之间,利用Lipofectamine 2000转染试剂(Invitrogen 11668019)将质粒转染到HeLa细胞中。转染24 h后,向培养基中添加终浓度为2 μg / mL的嘌呤霉素以进行筛选。24 h后弃去培养基,继续用含有2 μg / mL嘌呤霉素的完全培养基培养细胞,待培养皿中的单个细胞成长为大约20~40个细胞的克隆时,在显微镜下挑选单克隆至新的培养皿中。继续培养单克隆,在适当时候收集部分细胞,裂解后通过Western-blot进行基因敲低鉴定。
将收集到的细胞利用RIPA buffer裂解,通过Western-blot,以GAPDH为内参,利用抗USP16抗体对挑选出的细胞株系中USP16蛋白的表达量进行对比,筛选出了两株独立的usp16敲低细胞株,分别命名为KD-1和KD-3细胞。蛋白表达量使用Image J V1.53 h进行定量分析。
1.2.3 细胞增殖测定
将HeLa,KD-1和KD-3细胞分别接种每孔5×104个细胞于六孔板中,每隔24 h取一孔中的细胞,通过Trypan Blue法统计细胞数,持续6 d。使用R语言ggplot2包绘制细胞生长增殖折线图。
1.2.4 细胞内定位鉴定
将pcDNA3-RFP-USP16质粒在E.coliDH5α菌种中扩增培养后纯化质粒。待六孔板中细胞密度为70%~90%之间,利用Lipofectamine 2000(Invitrogen 11668019)转染试剂将质粒转染到293T细胞中。转染12 h后,利用0.25%胰蛋白酶将细胞分散,将1/10加入放置了干净细胞爬片的六孔板中,12 h后弃去培养基,PBS(NaCl 137 mmol/L,KCl 2.7 mmol/L,Na2HPO44.3 mmol/L,KH2PO41.4 mmol/L的水溶液)清洗两次,用4%多聚甲醛(Thermo 169650010)固定15 min,PBS清洗两次,将爬片放置到滴有DAPI染色剂(碧云天P0131)的载玻片上。使用显微镜(Leica DMi8)观察载玻片,并使用软件(LAS)在不同通道下拍照保存并合成。
1.2.5 细胞收集
将HeLa,KD-1,KD-3细胞在10 cm细胞培养皿中进行培养。每一个培养皿收集细胞为一个样本,每种细胞独立收集3个样本进行实验,即转录组深度测序和蛋白质组质谱实验中分别收集了9个样品进行了实验。细胞密度生长到90 %之上,使用细胞刮刀将细胞从培养皿底部刮下,用无菌PBS清洗3次。
1.2.6 转录组测序和数据处理
用于转录组深度测序的样本在用PBS清洗后,加入1 mL TRIzol(Tiangen DP424)在冰上吹打使细胞裂解,转移到细胞冻存管中,放入液氮中速冻30 min。速冻后从液氮中取出,放入80 ℃冰箱保存之后送上海美吉生物有限公司进行文库构建和深度测序。
采用RNA提取试剂盒(Invitrogen 15596026)提取样本中的总RNA,并使用DNase Ⅰ(TaKaRa 2270B)去除基因组DNA。使用ND-2000(NanoDrop Technologies)方法检测RNA的质量。取OD260/ OD280=1.8~2.2,OD260/ OD230≥2.0,RNA完整性值(RNA Integrity Number, RIN)≥6.5,m(28S RNA)/m(18S RNA)≥1.0,质量大于1 μg的RNA样本进行文库构建。采用TruSeqTM RNA sample preparation Kit(Illumina RS-122-2001)试剂盒构建文库,经TBS380(Picogreen)定量后,使用Illumina HiSeq Xten / NovaSeq 6000测序平台进行高通量测序,测序读长为PE150。
所得数据经SeqPrep(https:∥github.com/jstjohn/SeqPrep)和Sickle(https:∥github.com / najoshi/sickle)进行质控。使用HiSat2(http:∥ccb.jhu.edu/software/hisat2)与TopHat2(http:∥ccb.jhu.edu/software/tophat)将测序数据与人类基因组GRCh38.p13(http:∥asia.ensembl.org/Homo_sapiens/Info/Index)进行对比,通过RSEM(http:∥deweylab.github.io/RSEM/)计算基因的表达水平(TPM/FPKM)。基因表达差异通过Bioconductor中的edgeR包(http:∥www.bioconductor.org/packages/2.12/bioc/html/edgeR.html)和DESeq2包(http:∥bioconductor.org/packages/stats/bioc/DESeq2/)进行计算。当P<0.05且log2FC>1时,认为基因表达上调,当P<0.05且log2FC<-1时,认为基因表达下调。
1.2.7 蛋白质组质谱和数据处理
用于质谱蛋白质组学分析的样本在用PBS清洗后,使用含有8 mol/L盐酸胍和100 mmol/L Tris-HCl,pH8.0的裂解液,超声提取蛋白,并使用Bradford方法测定蛋白质浓度。加入裂解液将样品稀释至1 g/L,随后加入10 mmol/L DTT在37 ℃进行还原打开二硫键;还原后将样品取出放至室温后,加入100 mmol/L丙烯酰胺进行烷基化。还原烷基化后的样品使用超滤管置换成酶切缓冲液(50 mmol/L Tris-HCl,pH8.0),按照酶∶样品=1∶50的量先加入Lys-C,37 ℃条件下酶切2 h后,再按照相同比例加入胰蛋白酶,37 ℃消化过夜。收集酶切后的肽段冻干,并使用1%甲酸重溶。LC-MS/MS分析使用纳升液相EASY-nLC 1200串联Orbitrap Fusion Lumos质谱(Thermo Fisher Scientific),数据使用Proteome Discoverer(V2.4, Thermo Fisher Scientific)进行分析,搜库引擎为Mascot server(V2.7, Matrix Science)。Label-free定量使用内置LFQ搜库模板进行搜索,符合FDR(False Discovery Rate)≤0.01的结果被用来后续分析。当蛋白表达量改变P<0.05且log2FC>1时,认为蛋白表达上调,当蛋白表达量改变P<0.05且log2FC<-1时,认为蛋白表达下调。
1.2.8 差异基因功能分析
常规数据计算通过Excel和R语言完成,柱状图,火山图及条形图绘制使用ggplot2包,热图绘制使用pheatmap包,在将基因或蛋白的表达量进行归一化之后,使用k-means算法进行聚类分析,通过euclidean算法计算距离。获得基因或蛋白质显著上调和下调的列表后,通过Metascape[29](https:∥metascape.org/)将转录组和蛋白质组数据分析得到的列表进行整合,对其中共有的部分进行基因功能富集和蛋白互作网络分析,得到的蛋白互作网络文件使用Cytoscape V3.7.2打开,并绘制蛋白网络图,蛋白亚网络和关键节点由Metascape通过MCODE包自动进行计算分析。
2 结果与分析
2.1 检测usp16敲低细胞株的敲低效率
我们首先重复了USP16细胞内定位试验,通过将pcDNA3-RFP-USP16质粒表达于293T细胞中,以荧光信号定位USP16在细胞内的位置,确认USP16主要定位于细胞质中(图1(a),见第274页),与之前的研究一致[27]。组学分析需要稳定细胞株以获得可信的结果。为了增加结果的可信度,我们筛选出了来自两个单独克隆的两株usp16稳定敲低细胞株。在HeLa细胞中通过表达pHTP-shRNA-USP16质粒表达靶向usp16的shRNA,通过嘌呤霉素筛选清除不表达该靶向shRNA的细胞,再通过Western-blot,以GAPDH为内参检测USP16蛋白表达量降低的程度,最终我们挑选出两株USP16蛋白表达量下降明显的细胞株进行后续的试验,命名为KD-1和KD-3.USP16的表达量在KD-1和KD-3细胞中明显下降(图1(b),见第274页)。使用Image J V1.53 h对Western-blot结果进行定量分析,以USP16的蛋白表达量除去GAPDH的蛋白表达量得到USP16实际表达量,再将KD-1和KD-3细胞中USP16的蛋白表达量与HeLa对比得到相对表达量,我们确认usp16的敲低效率超过80%(图1(c),见第274页)。

图1 USP16细胞内定位与稳定敲低细胞株鉴定Fig.1 Subcellular location of USP16 and identification of usp16 knock-down(a) USP16主要定位于细胞质中,展示代表性图像;(b) Western blot检测稳定细胞株中USP16的表达量; (c) 稳定敲低细胞株中USP16的相对表达量。
2.2 usp16敲低导致细胞增殖减缓以及细胞形态发生明显变化
获得两株稳定usp16敲低细胞系KD-1和KD-3后,观察它们是否有明显的表型变化。根据之前的研究,USP16调控细胞增殖[21],我们检测了HeLa,KD-1和KD-3细胞的细胞增殖速度。USP16表达量的下降明显减缓了细胞的增殖速度(图2(a),见第274页)。有趣的是,KD-1和KD-3细胞的形态也发生了明显的改变,细胞延长为条状,伸出长突起(图2(b),见第274页)。结合USP16对线粒体-微管连接的形成以及染色体排布的调控作用,我们意识到USP16很可能通过对细胞骨架的调控,在细胞形态构建过程中发挥了重要作用。
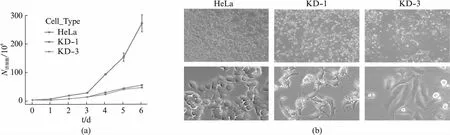
图2 usp16稳定敲低细胞系的细胞增殖和细胞形态发生明显改变Fig.2 Change of cell proliferation and morphogenesis of usp16 knock-down cells(a) KD-1与KD-3细胞的增殖速度明显慢于HeLa细胞;(b) KD-1和KD-3细胞的形态较HeLa细胞发生了明显的改变, 上方展示超过200个细胞,下方展示代表性图像。
2.3 usp16敲低细胞株的转录组数据比较分析
usp16敲低细胞系的细胞增殖和细胞形态都发生了明显的变化,我们通过转录组高通量测序,分别获得了HeLa,KD-1和KD-3细胞的转录组数据。分析发现,两株独立细胞系中基因的表达水平变化较为一致,证明了我们的敲低试验可靠且可重复(图3(a)),本研究中9个样本质控后共获得67.25 GB数据,比对率从88.32%到91.99%不等,共检测到表达基因32 435个,其中已知基因32 171个,新基因264个。在KD-1和KD-3细胞中,分别检测到3 295和3 285个基因的表达水平显著上调(P<0.05且log2FC>1),2 524和2 074个基因的表达水平显著下调(P<0.05且log2FC<-1)(图3(b,c))。共计2 497个基因在KD-1和KD-3中都明显上调,1 761个基因在KD-1和KD-3中都明显下调(图3(d))。这些在KD-1和KD-3中共同变化的基因受到USP16的明显调控。我们分别对上调和下调的基因进行了功能富集分析。
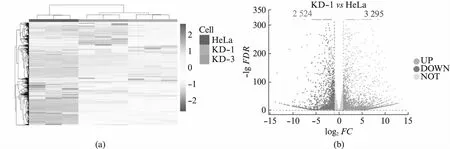
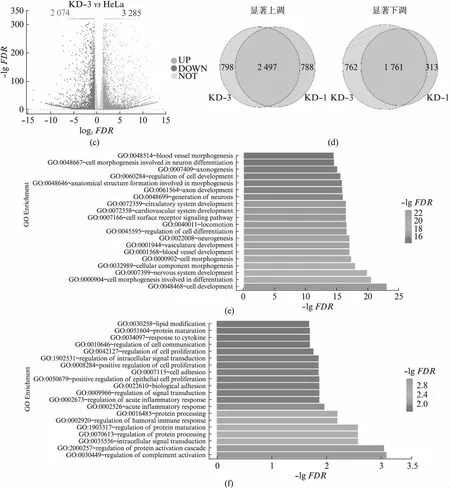
图3 usp16稳定敲低细胞系的转录组数据分析Fig.3 Transcriptome analysis of usp16 knock-down stable cell lines(a) 基因表达量分析热图;(b) 差异基因分析火山图,红色代表显著上调,蓝色代表显著下调,灰色代表不显著;(c) 不同细胞株系间 差异基因的对比;(d) 共同显著上调基因的功能富集分析;(e) 共同显著下调基因的功能富集分析。
上调的基因显著富集于细胞发育过程中,主要与细胞形态建成有关(图3(e))。细胞形态建成与神经系统发育,血管发育等过程密切相关[30],这些过程也表现出了明显的富集。USP16早先被发现与唐氏综合征小鼠的中枢神经系统干细胞受损的自我更新能力有关,我们意识到很可能这种关联是与USP16对细胞形态的调节有关[23]。细胞形态建成如轴突的建立对神经细胞发挥功能至关重要。考虑到USP16表达量下降后细胞明显拉长,后续的研究中有必要对比神经细胞与其他细胞中USP16的表达量是否有明显差异。下调的基因显著富集于与细胞膜活动相关的过程中,如补体活化、信号转导、免疫响应等(图3(f)),细胞形态的变化对于这些过程也是至关重要的,后续的研究有必要探究USP16在这些过程中的分子机制。
2.4 usp16敲低细胞株的蛋白质组数据比较分析
USP16调控了大量基因的表达,但是这种mRNA水平上的变化是否真实地体现在了蛋白质表达水平上,则需要通过质谱手段进行分析。分析发现,两株独立细胞系中蛋白表达量水平的变化较为一致,证明了我们的敲低试验可靠且可重复(图4(a),见第277页)。本研究中共检测到5 696个蛋白质。在KD-1和KD-3细胞中,分别有506和405种蛋白的表达水平显著上调(P<0.05且log2FC>1),499和481种蛋白的表达水平显著下调(P<0.05且log2FC<-1)(图4(b,c))。共计253种蛋白在KD-1和KD-3中都明显上调,323种蛋白在KD-1和KD-3中都明显下调(图4(d))。这些在KD-1和KD-3中共同变化的基因的蛋白水平受到USP16的明显调控。我们分别对上调和下调的蛋白进行了功能富集分析。上调的蛋白显著富集于细胞外结构组织、细胞-底物黏附、肌动蛋白结构组织等与细胞骨架构建相关的生物学过程中。这些过程均与细胞形态的改变有密切的关系,与转录组数据的结果是一致的(图4(e))。下调蛋白显著富集于线粒体中的氧化还原反应、线粒体排布、细胞内蛋白定位等过程。这些数据与转录组数据一致,表明USP16调控的蛋白主要与细胞骨架的构建,线粒体功能等生物学过程有关(图4(f))。
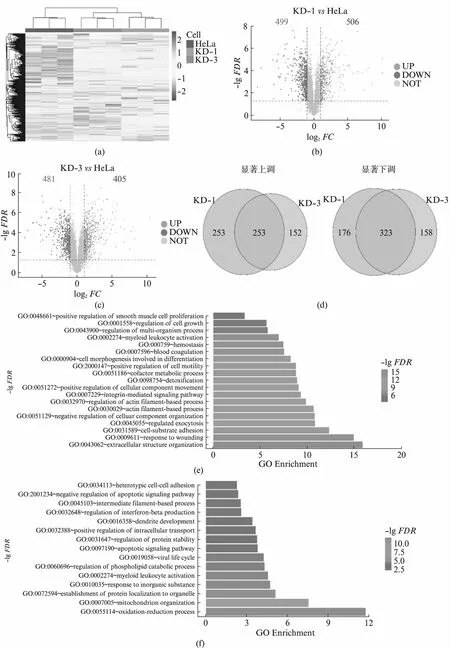
图4 usp16稳定敲低细胞系的蛋白质组学数据分析Fig.4 Proteomome analysis of usp16 knock-down stable cell lines(a) 蛋白表达量分析热图;(b、c) 差异蛋白分析火山图,红色代表显著上调,蓝色代表显著下调,灰色代表不显著;(d) 不同细胞株系间 差异蛋白的对比;(e) 共同显著上调蛋白的功能富集分析;(f) 共同显著下调蛋白的功能富集分析。
2.5 usp16稳定敲低细胞株的转录组和蛋白质组整合分析
令人惊讶的是,蛋白质组被调控的基因数量远远小于转录组水平被调控的基因的数量,这暗示了细胞内存在某种补偿机制能够对USP16的功能损失进行补偿,以维持细胞的各项生命活动。转录过程后细胞通过多种调节机制如表观遗传,蛋白修饰和降解等缓解usp16敲低对细胞造成的影响。我们对转录组中显著上调的基因进行类别分析,发现其中大约15%的基因编码lncRNA,显著下调的基因中,有大约20%编码lncRNA(图5(a),见第278页),这暗示了USP16通过表观遗传机制调控生命活动的其他作用。
转录组和蛋白质组的数据分析表明USP16调控了许多与细胞骨架建成,线粒体功能相关的基因,这在外表现为细胞形态发生明显变化、生长速度明显减缓,在内表现为大量与细胞骨架、细胞形态和线粒体功能相关的基因受到调控。转录组和蛋白质组数据中变化趋势一致的基因及其编码的蛋白受到USP16较为严格的调控,与USP16的关系更密切。为了了解具体是哪些基因和蛋白在USP16下游发挥重要功能,我们分别将显著上调或下调的转录组和蛋白质组列表进行整合的蛋白质互作网络分析(图5(b,c),见第278页),并计算网络中的关键节点,为后续的研究提供重要参考。

图5 usp16稳定敲低细胞系的转录组和蛋白质组对比分析Fig.5 Comparative analysis of transcriptome and proteome data of usp16 knock-down cells(a) 转录组显著差异基因类别分析;(b) 显著上调基因的蛋白互组网络图;(c) 显著下调基因的蛋白互作网络图; (d) 显著上调网络中的关键亚网络;(e) 显著下调网络中的关键亚网络。
显著上调的蛋白互作网络中,我们鉴定到7个对整个蛋白互作网络至关重要的亚网络(图5(d),见第278页),这些亚网络所代表的分子机制将是后续研究的重点,USP16如何调控这些蛋白质及这些蛋白质代表的生物学过程值得深入研究。
第1个亚网络与细胞骨架的构建有关,ADD1是细胞骨架相关蛋白,作为编码肌动蛋白-血影蛋白网络的关键组成部分,其作用是限制肌动蛋白丝的快速生长[31],ADD1的过量表达会促进细胞形成富含肌动球蛋白的突起[32],这与我们观察到KD-1和KD-3细胞出现长突起的表型一致。DST编码细胞骨架接头蛋白,充当中间丝连接肌动蛋白和微管细胞骨架网络[33]。PRKCA是丝氨酸/ 苏氨酸蛋白激酶,有许多蛋白质可以被其磷酸化,它可以直接磷酸化靶蛋白或者通过级联反应磷酸化下游蛋白质[34]。FLNC是肌肉特异的纤维蛋白,通过交联肌动蛋白发挥作用和参与信号响应中的肌动蛋白重组时间调控正常肌肉生成和维持肌肉纤维的结构完整性[35]。DST或FLNC的敲除会导致细胞突起减少,细胞增殖减缓,这与我们观察到KD-1和KD-3细胞的形态学变化和生长速度减缓一致,而PRKCA的过表达或敲除并不明显改变细胞形态。ITGA6是黏连蛋白的受体,在形成半桥粒的结构中发挥重要作用,它对细胞骨架的调控对IFG1和NRG1-ERBB等信号的转导是必不可少的[36],ITGA6的过表达增强了癌细胞的增殖能力,与我们观察到KD-1和KD-3细胞的增殖减缓一致[37]。形态观察和功能富集可以知道USP16调控了细胞骨架的构建和细胞形态建成,我们的研究表明USP16可能具体通过这些蛋白的表达,调控了相关的生物学过程,这为之后的研究指明了方向。
第2个亚网络与细胞周期中染色体的排布有关,CCNA是细胞周期蛋白,控制细胞周期从G1 / S向G2 / M过渡,它能够与细胞周期依赖性蛋白激酶CDK1和CDK2形成特定的复合物以激活它们,调控细胞周期[38],CCNA敲低会影响细胞周期进程,抑制癌细胞的生长,促进细胞凋亡[39]。这与前文提到的KD-1和KD-3细胞系的表型一致。CDK15编码细胞周期依赖的蛋白激酶[40],这一发现与早先USP16能够调控细胞周期的研究是一致的,且CDK15的敲低会导致癌细胞增殖能力减弱,与本文中的观察一致。之前的研究集中于CDK1而非CDK15(另一个同源的细胞周期依赖的蛋白激酶),且对USP16的调控网络并不了解[27],后续有必要对CDK15是否受到USP16的调控进行进一步的研究。H1-1和H1-3是组蛋白H1的两种亚型,分别与细胞周期和RNA结合等过程有关。UBASH3B是一种酪氨酸酶,对包括EGFR、FAK在内的大量蛋白有作用。后续研究有必要对我们新发现的USP16调控蛋白进行更深入的研究。
第3个亚网络与细胞黏附和物质代谢有关,ARSB为芳基硫酸酯酶B,它能从4-硫酸软骨素(C4S)中除去硫酸基团并调节其降解,通过参与细胞黏附调控细胞迁移和侵袭等过程。它是中枢神经系统中神经突生长和神经元可塑性的调节剂[41]。GLA是半乳糖苷酶,DPP7为寡肽水解酶,UNC13D和NPC2对胞吐作用有重要影响。
第4个亚网络与线粒体代谢相关,PRKAR1A是cAMP依赖性蛋白激酶Ⅰ型α亚基,调控cAMP信号转导[42]。ALDH1B1和ALDH2是线粒体中醛脱氢酶的主要组件,调控线粒体中的代谢活动[43]。HK1是己糖激酶1,催化细胞中各种己糖的磷酸化[44]。SDSL是丝氨酸和苏氨酸脱氢酶,参与各种代谢过程。
第5个亚网络与溶酶体功能有关,SLC44A2是胆碱转运蛋白[45]。PLAUR尿激酶纤溶酶原激活剂的受体,在定位和促进纤溶酶形成中发挥作用[46]。LAMTOR2是Ragulator复合物的组件,参与氨基酸信号转导和mTORC1信号的激活以激活溶酶体[47]。PGRMC1是孕激素结合蛋白复合物的组分,参与信号转导[48]。
第6个亚网络与泛素化相关,UBE2S是泛素结合酶E2S,它能够结合被E1激活的泛素蛋白,并将泛素通过E3转移给底物蛋白,它能够增强对细胞周期中发挥重要功能的APC / C(Anaphase Promoting Complex / Cyclosome)复合物的多泛素化,并导致其通过蛋白酶体途径降解,由此抑制有丝分裂[49]。HECTD1、MEX3C、TRIP12是3种E3泛素连接酶。值得注意的是,MEX3C是一种RNA结合的E3泛素连接酶,它能够直接与某些mRNA的3’UTR区域结合,阻止蛋白质的翻译,但相关的机制尚不清楚[50]。USP16作为一种泛素化酶,当其表达量下降时,这些与泛素化相关的蛋白质反而表达量升高,或许在细胞内作为某种反馈机制有所存在,但USP16发挥去除H2A单泛素化的功能,这需要我们进行更为深入的研究。
第7个亚网络与胞内运输有关,MAPRE3与微管蛋白结合,通过稳定微管促进细胞骨架的形成[51]。KIF3A和KIF3B组成异源三聚体,作为驱动蛋白发挥功能,在细胞内可以用于蛋白质复合物、核酸、细胞器的运输[52]。
这几个亚网络中,某些USP16调控的功能已经被发现,但也有许多尚未被研究,尤其是与细胞骨架构建和线粒体功能相关的蛋白质值得我们进行更进一步的研究。
显著下调的蛋白互作网络中,我们鉴定到16个关键亚网络(图5(e)),这些关键亚网络主要与蛋白质翻译、线粒体中蛋白质翻译、线粒体电子传导、线粒体物质代谢、线粒体物质运输等有关。
第1个亚网络与调控基因表达有关,与之前的发现一致[53-54],但我们发现了USP16对于基因转录的调控是透过调控INTS6,SIRT1,INTS3,MYC等的表达实现的,这一点尚未有相关的研究。
第2、3个网络分别与线粒体中的基因表达,线粒体中的物质运输有关,这一点非常有趣,USP16的研究一直集中于它作为组蛋白H2AK119ub去泛素化酶,早前的蛋白质组学研究通过分离细胞成分发现USP16也可能定位于线粒体中[55],但始终没有研究报道USP16是否在线粒体中发挥功能,USP16在线粒体中的底物是什么,以及在线粒体中发挥了什么功能。本文可以佐证之前的研究,即USP16可能在线粒体中发挥调控基因表达的功能,且我们发现了潜在的USP16下游的基因,如第2个亚网络中PTCD3与线粒体中的蛋白翻译有关[56],AUH与线粒体中甲基谷氨酰辅酶A的代谢有关[57],MRPS6,MRPS24,MRPS31,MRPS35是线粒体中核糖核蛋白的重要组成成分,与线粒体中的蛋白翻译有关[58]。TRUB2是核糖体中将mRNA假尿苷化的酶,参与核糖核蛋白体的构成,调控了蛋白翻译[59]。考虑到线粒体的进化来源和原核生物中不存在泛素化系统的事实,这样的发现非常有趣。第3个亚网络中,TMEM70与线粒体中ATP的合成有关[60],COA6,COX2,COX5B,COX6A1,COX6B1,COX6C是线粒体呼吸链的重要成分[61],usp16敲低细胞的生长减缓很可能与它的线粒体功能受损密切相关。usp16敲低细胞的增殖速度减缓与线粒体相关蛋白表达量的下调是一致的。USP16表达量下降后,细胞的代谢受到了明显的影响,在分子机制上表现为线粒体中的蛋白表达和能量代谢相关蛋白表达量下降,在细胞发育过程中表现为增殖减缓。考虑到USP16在造血功能受损、癌症发生、胚胎发育受阻等病理学过程中的重要作用,通过USP16可将细胞核内的表观遗传修饰与线粒体中的代谢活动联系起来。
第4个亚网络是核仁的重要组成成分,与rRNA的合成和修饰有关,UTP6,UTP14A,IMP3是U3 snoRNP的重要成分,对18S核糖体RNA的剪切有关[62-63],NOL12和NOP14等是核仁的重要蛋白部件,这样的发现暗示USP16可能通过调控核糖体的组建发挥功能,但目前缺乏相关的研究。考虑到癌细胞的增殖速度加快,代谢水平显著升高,usp16敲低细胞的增殖速度减慢,代谢相关的蛋白网络被下调,暗示usp16作为靶点在癌症治疗中有潜在的价值。
第5个亚网络与桥粒形成和稳定有关[64],这进一步确认了本文的研究。其他亚网络与mRNA出核,泛素化-蛋白酶体降解系统等有关。
3 讨 论
USP16是组蛋白H2AK119ub主要的去泛素化酶,调控了基因转录,细胞增殖,细胞分化,细胞周期,干细胞自我更新等重要的生物学过程[13,15,18-19,21,23,27-28,65]。长期以来,对USP16的研究都集中于它在细胞核中如何通过去泛素化H2AK119ub调节癌症发生、造血功能异常、胚胎发育停滞、神经系统受损等病理学过程。考虑到USP16被积极排除在细胞核外,只在有丝分裂G2 / M期间进入细胞核内发挥功能[22],缺乏对USP16调控的功能调控网络的整体性理解。本研究通过整合usp16稳定敲低细胞系的转录组和蛋白质组数据,对USP16调控的基因在转录和翻译层面进行了全面的了解。USP16的表达失控或者活性缺失对细胞是毒性的[20],由于USP16在转录层面和蛋白层面调控的基因数量差距较大,故细胞中可能存在某种补偿机制或抑制机制抵消usp16敲低对细胞带来的影响。
在本研究中,我们构建并筛选usp16稳定敲低的细胞株,与HeLa细胞对比发现其生长速度和细胞形态上的明显变化。我们通过分析转录组深度测序数据和蛋白质组学数据,了解了USP16具体调控了哪些生物学过程。usp16稳定敲低细胞系明显的形态变化与转录组和蛋白质组数据互为佐证,且我们发现这种调控是透过对细胞骨架的调节实现的。相关的蛋白网络在usp16敲低时明显上调,但具体的分子机制仍需要进一步深入研究。USP16与细胞形态调控的关系值得在细胞形态变化较大的细胞,如骨骼肌细胞、神经细胞、免疫细胞等中进行研究。USP16与唐氏综合征有关,但是具体的分子机制并不为人所知,如果能够探究USP16是否调控神经细胞的发育,将为相关疾病的治疗提供帮助。
之前的蛋白质组学研究发现USP16可能在线粒体中发挥功能,本文发现USP16调控了线粒体中关于蛋白质翻译和细胞呼吸链相关的蛋白网络,考虑到线粒体的进化来源和功能特点,这样的发现为阐明线粒体与细胞核的互动提供了理论依据。后续的研究应当探究USP16在细胞核,细胞质,线粒体等中的定位是否会在细胞发育,癌症发生等过程中发生变化,研究USP16在线粒体中的功能会将经典的表观遗传学现象和细胞代谢联系起来,具有重要的生物学意义。例如,USP16与线粒体功能的关系可以在心脏模型,骨骼肌模型等需要消耗大量能量的过程中进行分析。
USP16与PRC1通常被认为拮抗调控细胞的生长发育,我们认为有必要也对PRC1进行类似的研究,即通过敲低PRC1,研究下游的基因和调控网络。PRC1的催化核心由RING1A、RING1B和BMI组成,其中RING1B的作用最重要。我们发现还没有人做过类似的分析。先敲低RING1B,我们能够了解PRC1是否对细胞形态和线粒体功能有调节作用。更进一步,我们还能够将USP16和PRC1的调控网络进行对比分析,对USP16和PRC1的功能进行细致的研究。两者之间不同的调控网络可能会揭示USP16与PRC1尚未报道的新功能或调控作用。
猜你喜欢
重庆理工大学学报(自然科学)(2022年9期)2022-10-26
中国药理学与毒理学杂志(2022年7期)2022-10-17
检验医学与临床(2022年19期)2022-10-10
中华实用诊断与治疗杂志(2022年1期)2022-08-31
生物化学与生物物理进展(2022年8期)2022-08-20
科学导报(2022年44期)2022-07-25
中国医药导报(2019年13期)2019-06-20
分析化学(2019年3期)2019-03-30
中国现代医生(2018年22期)2018-12-04
飞碟探索(2016年4期)2016-04-07
