
铁系无机材料用于新能源电催化硝酸盐还原制氨*
2022-07-02 03:17任柯丞许彦桐
新能源进展 2022年3期
任柯丞,许彦桐,曹 晏
铁系无机材料用于新能源电催化硝酸盐还原制氨*
任柯丞1,2,3#,许彦桐2,3#,曹 晏1,2,3†
(1. 安徽大学 化学化工学院,合肥 230601;2. 中国科学院广州能源研究所,广州 510640;3. 广东省新能源和可再生能源研究开发与应用重点实验室,广州 510640)
硝酸盐是一类普遍存在的环境污染物,而其对应的还原态氨却是重要的化工原料和农业肥料。因此,探索将两者直接结合的化学转化具有重大的技术及经济战略意义,尤其是对当今“双碳”战略驱动下的高碳排放合成氨的工艺变革以及氨可能成为下一代载氢燃料。通过电催化技术,将硝酸盐还原合成氨过程与可再生能源电力结合,构建绿色低碳的含氮化学品人工“氮循环”的新循环技术与经济体系,是解决目前合成氨工业对化石能源高度依赖、高碳排放问题以及开发新氢能的有力途径。借鉴传统合成氨催化广泛应用、具有成本优势的铁系催化机制,分别选取单质Fe、Fe2O3和Fe3O4作为电催化材料,探索并揭示电催化硝酸盐还原合成氨催化反应的化学形态。结果表明:在相对于可逆氢电极电势为−0.53 ~ −0.93 V区间内,Fe2O3表现出了最优异的催化活性,其生成氨的法拉第效率(FENH3)最高可达88%,对应的生成氨电流密度(NH3)为43.1 mA/cm2、氨生成速率(NH3)为0.20 mmol/(cm2·h)。此外,从控制硝酸盐转化率选择性获得氨和硝酸铵两种不同产物的技术路线,分析对比了相应的用电成本以及产品市场价格,充分说明了路线的经济性,说明铁系无机材料在电催化硝酸盐还原合成氨方面具有非常巨大的市场化潜力。
铁系材料;硝酸盐还原;合成氨;电催化
0 引 言
氨是最重要的基础化工原料之一,被广泛用于肥料、国防、制药、染料和合成纤维等领域[1]。其中,全球将近80%的氨被用于化肥行业,对现代农业以及维持全球人口增长起关键作用[2]。此外,液氨也是一种重要氢能载体,其体积能量密度是液氢的1.53倍,具有储氢量大、能量密度高和易于运输储存的优势[3-4]。因此,将液氨直接作为化学燃料,具有高燃烧热值、无温室气体排放和绿色环保等优点,有望成为未来最重要的可持续清洁能源之一[5-6]。但在“双碳”战略背景下,许多传统行业都必将迎来一轮产业改革和落后产能淘汰,而其中也包括了高能耗和高碳排放的传统合成氨工业。目前合成氨主要依赖于哈伯−博施法(Haber-Bosch process)[7],即氢气和氮气经铁触媒催化,在高温(350 ~ 500℃)、高压(150 ~ 300 bar)条件下反应得到[8]。而这个过程中的原料氢气和氮气的生产、提纯和压缩等过程均高度依赖化石能源[9-10]。寻找新型的“低碳”,甚至“零碳”合成氨工艺成为接下来合成氨工业升级的当务之急[11-12]。
近年来,以含氮化学品为代表的各种化学污染物对地下水和土壤的污染引起了广泛关注[13]。这些含氮化合物可能导致水富营养化并进一步污染人类饮用水[14];并且饮用水中硝酸盐含量超标也是高铁血红蛋白血症和高癌症发病率的主要原因[15-16]。世界卫生组织建议饮用水中的硝酸盐含量应低于50 mg/L(约11.3 mg-N/L),亚硝酸盐含量应低于3 mg/L(约0.9 mg-N/L)[17]。同时,由于含硝酸盐废水来源广泛(包括农业、工业和生活废水等)、分布式排放、排放量大等特点,不仅对生态环境构成严重威胁,而且对人类的健康发展也有许多不利影响,监控、回收并转化污水中硝酸盐一直是环保行业的关注重点之一[18-19]。针对废水硝酸盐的处理,人们已经开发了反渗透[20]、离子交换[21-22]和细菌反硝化[23]等技术手段来转化硝酸盐[24]。然而,这些传统脱硝技术也存在着高能耗、产生附加经济价值低等缺点难以在“双碳”战略框架下可持续发展[25]。
近年来,有学者提出了电催化硝酸盐还原制氨(nitrate-to-ammonia reduction reaction, NARR)的新技术路线[26]。该技术路线可结合可再生能源发电技术,实现在常温常压条件下合成氨[27];同时直接利用H2O作为反应的氢源,避免了从化石燃料中获取H2,从原理上实现了“零碳”合成氨[28]。此外,该路线还可对接环保企业中的脱硝工艺,避免产生低价值、难以利用的N2,建立人工“氮循环”,实现含氮化学工业的可持续发展[29-30]。
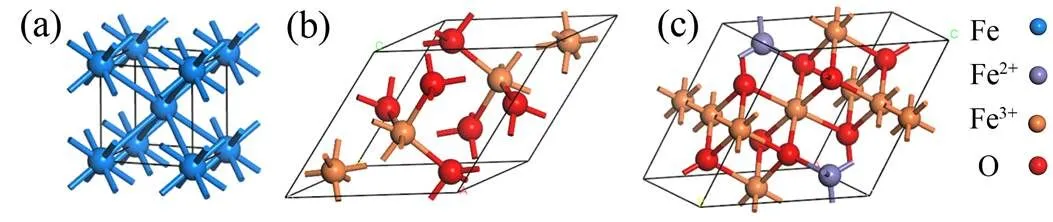
受启发于现有合成氨工艺中广泛应用的铁触媒催化剂,同时结合我国作为铁资源大国的基本国情[31],本文将探讨铁系无机材料中最重要的单质Fe、Fe2O3和Fe3O4(如图1所示)作为电催化硝酸盐还原合成氨催化剂的可行性:通过一系列的物理表征与电化学合成氨催化性能测试分析其技术可行性;再通过对产物价值以及用电成本分析其经济可行性,为设计高效、经济与“零碳”的新型合成氨工艺提供新的研究设计思路。
1 实验部分
1.1 试剂与仪器
采用的试剂四氧化三铁(Fe3O4, 97%)、硝酸钾(KNO3, ≥ 99%)、乙醇(99.8%)、磷酸二氢钾(KH2PO4, 99.5%)、磷酸氢二钾(K2HPO4, 99%)、氢氧化钾(KOH, 85%)、氯化铵(NH4Cl, 99.999%)均购自阿拉丁试剂(上海)有限公司;碳布(W1S1009, 0.41 mm)购自碳能科技股份公司;氩气(99.999%)购自广州盛盈化工有限公司。
实验仪器包括X’Pert PRO MPD型多晶粉末X射线衍射仪(X-ray diffractometer, XRD)、日立S-4800型冷场发射扫描电镜(scanning electron microscope, SEM)、上海辰华CHI760E电化学工作站、德国Fritsch PULVERISETTE 7小型行星式球磨机。
1.2 催化剂的制备
1.2.1 Fe3O4粉末的制备
将市售Fe3O4放入行星高能球磨机中以800 r/min的转速球磨40 min,获得粒径小于500 nm的Fe3O4。
1.2.2 单质Fe粉末的制备
称量1 g球磨处理后的Fe3O4粉末作为原料,在10% H2的Ar-H2气氛下(30 mL/min),以10℃/min升温至350℃,随后保持12 h,冷却至室温后将样品转移至行星高能球磨机中以800 r/min球磨10 min,得到纯的且粒径均匀的单质Fe粉末。
1.2.3 Fe2O3粉末的制备
称取1 g球磨处理后的Fe3O4粉末作为原料,在空气氛围中,以10℃/min升温至500℃,并保持12 h后,冷却至室温后继续放入行星高能球磨机中以800 r/min球磨10 min,得到纯的且粒径均匀的Fe2O3粉末。
1.3 实验准备
1.3.1 电极的制备
分别准确称取10 mg Fe, Fe2O3和Fe3O4催化剂粉末加入试管中,加入0.4 mL去离子水、0.8 mL无水乙醇和40 μL的全氟磺酸型聚合物溶液(Nafion,质量分数为5%),放入超声清洗机中超声分散1 h,使团聚在一起的Fe2O3充分分散并形成均一稳定的悬浊液。
将1 cm × 2 cm的碳布依次在无水乙醇和去离子水中超声各处理1 h后,自然晾干。随后将处理好的催化剂悬浊液滴在碳布上面,并通过称重法确保三种催化剂的Fe元素负载量为22 μmol/cm2。需指出的是制备单质Fe的电极时,电极干燥过程采用Ar气进行保护,以避免单质Fe被氧化。
1.3.2 质子交换膜活化
将市售杜邦Nafion 211质子交换膜置于稀硫酸(质量浓度为5%)中,并在80℃下保存0.5 h;随后用大量的去离子水洗涤若干次,并在去离子水中浸泡3 h后取出备用。
1.3.3 KNO3电解液的配制
称取0.25 mol的KH2PO4、0.25 mol的K2HPO4和0.5 mol的KNO3,加入去离子水将其溶解并转移至500 mL容量瓶中定容,配制成NO3−浓度为1.0 mol/L的电解液(pH = 6.6)。所有电化学实验开始前,均预先往电解液中通入高纯Ar至少30 min。
含0.5 mol/L和1.5 mol/L的KNO3电解液均依照此方法加入对应剂量的KNO3进行配制,KH2PO4和K2HPO4浓度不变。
1.3.4 NH3/NH4+浓度标准曲线
配制0.2 mmol/L、1 mmol/L、2 mmol/L和4 mmol/L的NH4Cl溶液作为标准溶液,通过氨氮分析仪检测记录相应的吸光度,线性拟合计算出氨浓度标准曲线图2。
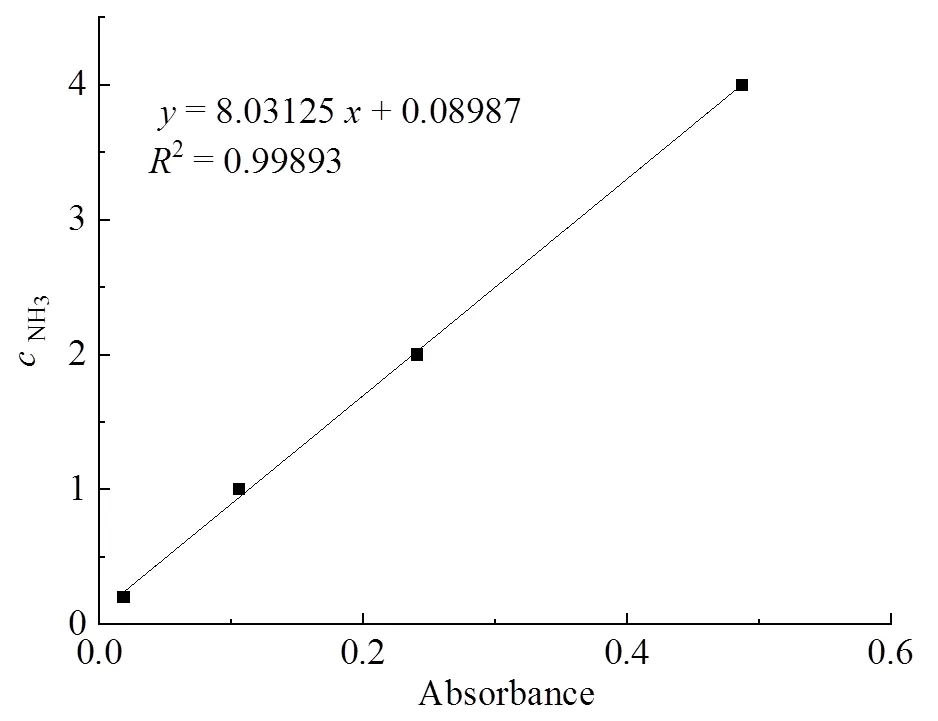
图2 NH3/NH4+ 摩尔浓度的标准曲线
1.4 电化学实验
所有电化学测试均在H型电解池中进行,并使用质子交换膜对阴极和阳极进行分隔,如图3所示。在阳极池和阴极池中分别加入30 mL的电解液。电化学测量采用三电极法,其中以高纯石墨棒作为对电极,以饱和KCl溶液填充的Ag/AgCl电极为参比电极(Ag/AgCl= 0.197 V,相对于标准氢电极电势),用负载催化剂的碳布作为工作电极。本文所涉及的电极电势均以可逆氢电极(reversible hydrogen electrode, RHE)为参考,相关电位可利用能斯特方程计算得到RHE=exp+ 0.059pH +Ag/AgCl,其中exp为实验测试所得。

图3 电解池示意图
线性扫描伏安(linear sweep voltammetry, LSV)测试的扫描电势区间为−0.4 ~ −1.2 V,扫描速度为10 mV/s,所有LSV曲线均未进行补偿。
循环伏安(cyclic voltammetry, CV)曲线测试过程中不断向电解液通入50 mL/min的高纯Ar,电势扫描区间为−0.1 ~ 0.1 V (相对于RHE),扫描速率分别为10 mV/s、30 mV/s、50 mV/s和70 mV/s。取CV曲线中阳极电流与阴极电流均值的差值作为扫描速率的函数,线性拟合确定材料的单位面积电容dl,通过文献给出的公式计算电化学活性面积(electrochemical active surface area, ECSA)[32]。
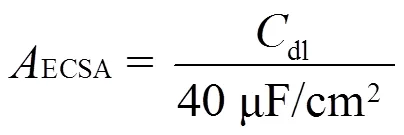
电催化实验在密封情况下进行,分别在−0.53 V、−0.63 V、−0.73 V、−0.83 V和−0.93 V(相对于RHE)的电位下,持续电解30 min。利用氨氮分析仪对电解后溶液中的NH3浓度进行分析,记录对应吸光度数值。通过如图2所示的标准曲线计算获得对应的NH3浓度,进而分别计算得出生成NH3的法拉第效率(FENH3)、氨电流密度(NH3)和氨生成速率(NH3):
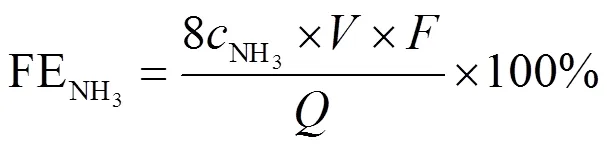

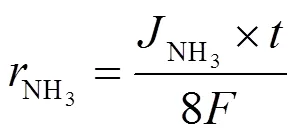
其中:是法拉第常数,96 485 C/mol;为阴极室电解液体积,30 mL;NH3为测得的NH3浓度,mol/L;为总电荷量,C;为碳布面积,2 cm2;为电解时间,s。
-曲线与循环稳定性测试方法为在−0.83 V下电解6 h后,收集分析电解液中的NH3浓度,并更换电解液,继续进行持续6 h的循环测试,合计循环4次。
1.5 经济分析中的成本计算
NH3和NH4NO3的生产路线对应的每吨产品用电成本NH3和NH4NO3分别通过以下公式进行计算:
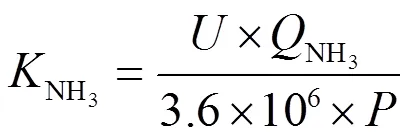
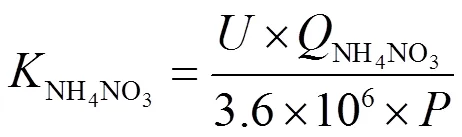
式中:NH3和NH4NO3分别为生产1 t NH3和NH4NO3成本;为电解电压;NH3是生产1 t NH3所需要的总电荷;NH4NO3是生产 1 t NH4NO3所需要的总电荷;为相应用电类型每度电的电价。
2 结果与讨论
2.1 硝酸根的还原反应
硝酸盐还原合成氨反应是一个八电子还原过程[33-34]。虽然其热力学可行性已被充分证明,但是在其高选择性与高反应动力学控制方面却依然存在着许多挑战[35]。由于硝酸盐的低电荷密度和离域化的电子结构以及阴离子特性,使其与阴极催化剂的结合力比起水溶液中其他分子或离子(H2O、H+等)要弱得多[36]。此外,NO3−可以逐级获得电子而被还原为不同产物,且不同产物的热力学还原电位非常接近。因此在NO3−还原合成氨过程中极有可能出现多种还原产物(如图4所示),精准控制反应过程的电子转移数与转移速率,以获得高选择性与高反应速率的NH3产物非常困难[37]。
通常认为亚硝酸盐形成步骤是NARR反应中的限速步骤之一,且在反应的动力学中起着决定性的作用。结合文献调研[38],NO3−在铁原子催化作用下的还原过程极有可能经历吸附、脱氧、加氢、脱附的变化过程(NO3−→ *NO3→ *NO2→ *NO → *NHO→*NH2O→*NH2OH→*NH2→*NH3→NH3)。
2.2 单质Fe、Fe2O3和Fe3O4的表征
所使用的单质Fe、Fe2O3和Fe3O4的XRD图谱如图5所示。结合数据库中标准的PDF卡片(Fe: 87-0722; Fe2O3: 72-0469; Fe3O4: 88-0315)可以得出结论:(1)三份样品的衍射峰均较窄,说明均有较高的结晶度;(2)没有多余的杂峰,尤其是在单质Fe和Fe2O3中未观察到Fe3O4衍射峰,说明原料Fe3O4已完全转化,所使用催化剂具有较高纯度。另外,从样品的SEM图(图6a ~ 图6c)图可以看出,三种材料均为纳米级球形颗粒,表面均匀,粒径均保持在70 nm左右(图6d ~ 图6f)。三份样品形貌尺寸差异不大,在一定程度上可以排除介观尺度上反应物在催化剂表面微区传质对性能产生影响,后续NARR电催化实验中表现出的性能差异主要是由物质本身引起的。
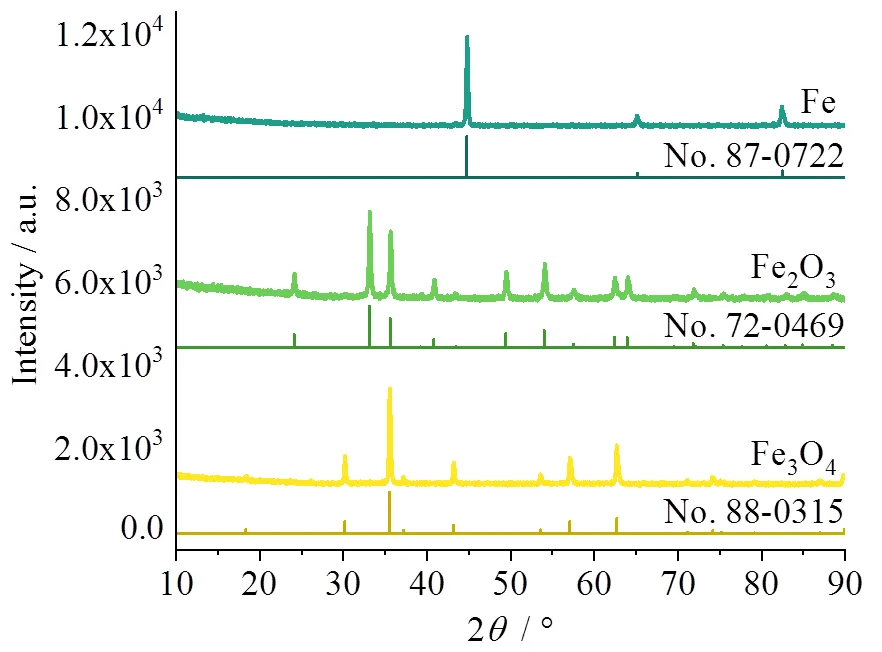
图5 单质Fe、Fe2O3和Fe3O4的XRD图

2.3 电催化NARR性能测试
2.3.1 电解实验条件优化
首先采用含有1.0 mol/L KNO3的磷酸盐缓冲溶液(phosphate buffered saline, PBS)以及空白溶液(即不含KNO3的PBS)分别作为电解液,以Fe3O4作为NARR的催化剂,进行LSV曲线测试,结果如图7a所示。在含有NO3−的电解液中测试所获得的电流明显高于空白溶液,表明NO3−在该电位区间可以发生电化学反应。另外,经过在−0.83 V下电解30 min后,两份电解液的NH3浓度差异很明显(如图7b):含硝酸盐的电解液对应的NH3浓度为2.82 mmol/L,而空白溶液的NH3浓度仅为0.04 mmol/L,考虑到环境影响以及实验误差,这个结果几乎可忽略不计。以上实验说明Fe3O4具有NARR电催化活性。
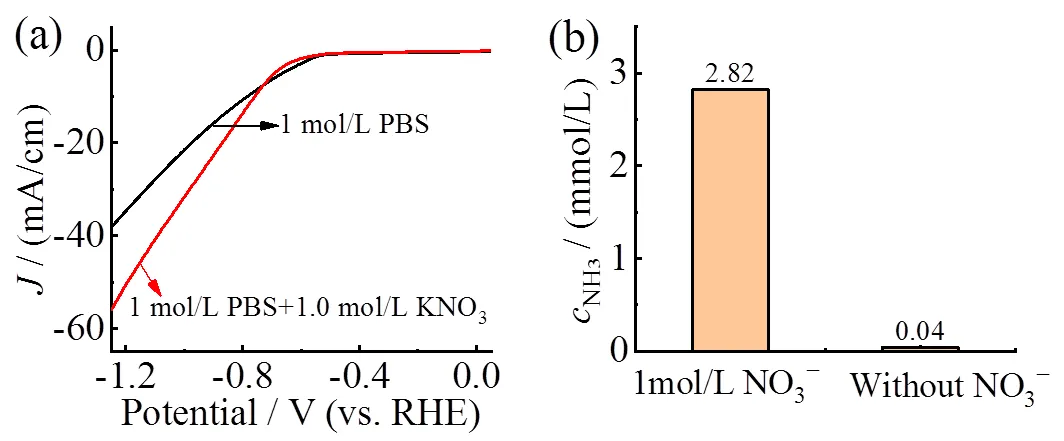
图7 Fe3O4在含有KNO3缓冲溶液和不含KNO3的缓冲溶液中的电化学性能对比:(a)LSV;(b)cNH3

图8 Fe3O4在不同浓度磷酸缓冲溶液中的法拉第效率对比
另外分别取含有0.5 mol/L、1.0 mol/L和1.5 mol/L KNO3的PBS作为电解液,同样以Fe3O4作为工作电极的催化剂,结合LSV测试结果,在−0.53 ~ −0.93 V的电势区间每隔0.1 V进行一次30 min的恒电压电解,并分析电解液中产出的NH3浓度,获得不同电势下对应生成氨的FENH3数据。实验结果如图8所示。在1.0 mol/L的NO3−电解液中的FENH3总体均略高于1.5 mol/L和0.5 mol/L KNO3磷酸缓冲溶液中的FENH3,说明1.0 mol/L为该体系的最优反应物浓度,此时电催化NO3−还原为NH3的选择性相对最高。故后续其他铁系催化剂的电催化性能评价实验也在该浓度下进行对比讨论。
2.3.2 不同铁系催化剂的电催化性能对比
单质Fe、Fe2O3和Fe3O4在含有1.0 mol/L KNO3的PBS中的LSV曲线对比结果如图9a所示。三种催化剂的反应起始电压均在−0.51 V(相对于RHE)附近(选取电流密度= 0.8 mA/cm2作为参考标准);但随着电压继续减小,其电流密度开始呈现出差异,总体规律是Fe2O3>Fe3O4>Fe,即这三种催化剂对NARR的电催化活性的顺序。
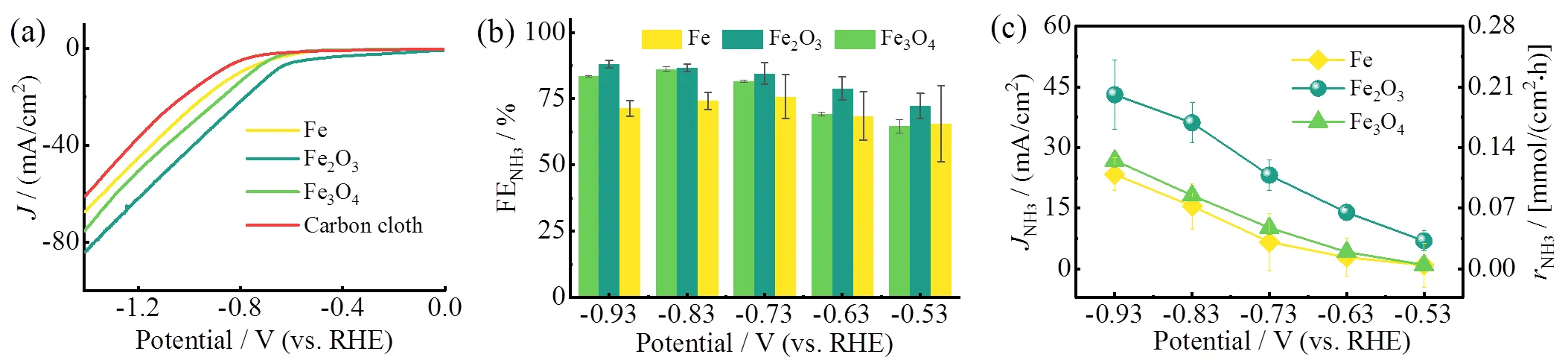
图9 三种铁系催化剂在1.0 mol/L NO3−电解液中的NARR性能对比:(a)LSV;(b)FENH3;(c)JNH3与对应的rNH3
随后分别以单质Fe、Fe2O3和Fe3O4为工作电极,在1.0 mol/L的NO3−电解液中采用不同电压进行30 min的恒电压电解,并定量分析催化反应后的电解液中产出的NH3浓度,计算出FENH3、NH3以及相应的NH3,结果如图9b和图9c所示。从图9b中可以看出,Fe2O3在−0.93 V时FENH3最高,达到了88%;而且三种铁系催化剂的FENH3均随反应电压的降低逐渐增大,最后趋于平稳。这可能是由于NARR为八电子还原过程,其电子转移需要克服更高的能垒,因此相对更负的电势更有利于多电子转移过程。另外,在电压达到−0.73 V(相对于RHE)时,Fe2O3和Fe3O4的FENH3分别稳定在84.5%和81.7%,远高于单质Fe的75.7%,随着电压进一步降低,FENH3趋于稳定。这意味着这三种催化剂在电催化NARR上均具有较宽的工作窗口,尤其是Fe2O3,可以使硝酸盐还原过程保持着较高的NH3选择性,有较大的应用潜力。
尽管Fe2O3和Fe3O4的FENH3相近,但通过图9c的NH3对比图看出,Fe2O3的NH3远大于Fe3O4和Fe。其中,Fe2O3在−0.93 V(相对于RHE)时的NH3可达43.1 mA/cm2,对应的NH3为0.20 mmol/(cm2·h);而Fe3O4和Fe仅分别为26.8 mA/cm2和23.3 mA/cm2,对应的NH3为0.13 mmol/(cm2·h)、0.11 mmol/(cm2·h)。说明Fe2O3在高FENH3的条件下可保持更高的产氨速率,进一步说明其巨大的应用潜力。
通过评估ECSA进一步探索三种铁系催化剂的活性根源,结果如图10所示。采用双电容层法(double layer capacitance, DLC)测试样品的ECSA。在扫描速度分别为10 mV/s、30 mV/s、50 mV/s和70 mV/s的条件下测定单质Fe、Fe2O3和Fe3O4的CV曲线(图10a ~ 图10c)。结果表明,实验所用的单质Fe、Fe2O3和Fe3O4电极的单位面积电容分别为0.292 mF/cm2、0.348 mF/cm2和0.352 mF/cm2,对应的ECSA分别为7.30 cm2/cm2、8.70 cm2/cm2和8.80 cm2/cm2。从测试结果可以看出三者ECSA大小为Fe3O4> Fe2O3> Fe。基于ECSA计算−0.93 V下Fe2O3、Fe3O4和单质Fe对应的氨电流密度(NH3,ECSA)分别为4.95 mA/cm2、3.05 mA/cm2和4.24 mA/cm2。虽然Fe2O3的ECSA介于单质Fe和Fe3O4之间,但是其NH3,ECSA仍然大于Fe和Fe3O4,进一步表明Fe2O3的本征催化活性是三者中最高的。
三种催化剂NARR活性的差异很可能与铁原子价态有关。如前所述,硝酸根的还原首先需要经历硝酸根的吸附,而带负电荷的硝酸根在铁原子上的吸附强度应该与铁原子表面的电荷密度相关,铁原子正电荷密度越大,吸附越强。因此,Fe(III)有利于促进硝酸根的吸附,即Fe2O3活性最高。而Fe3O4虽然也含有Fe(III),但在铁位点数量保持相同的前提下,部分Fe(III)被Fe(II)替代,因此活性有所降低。而单质铁由于只含Fe(0),正电荷密度最低,最不利于硝酸根吸附,因此宏观的NARR活性最差。
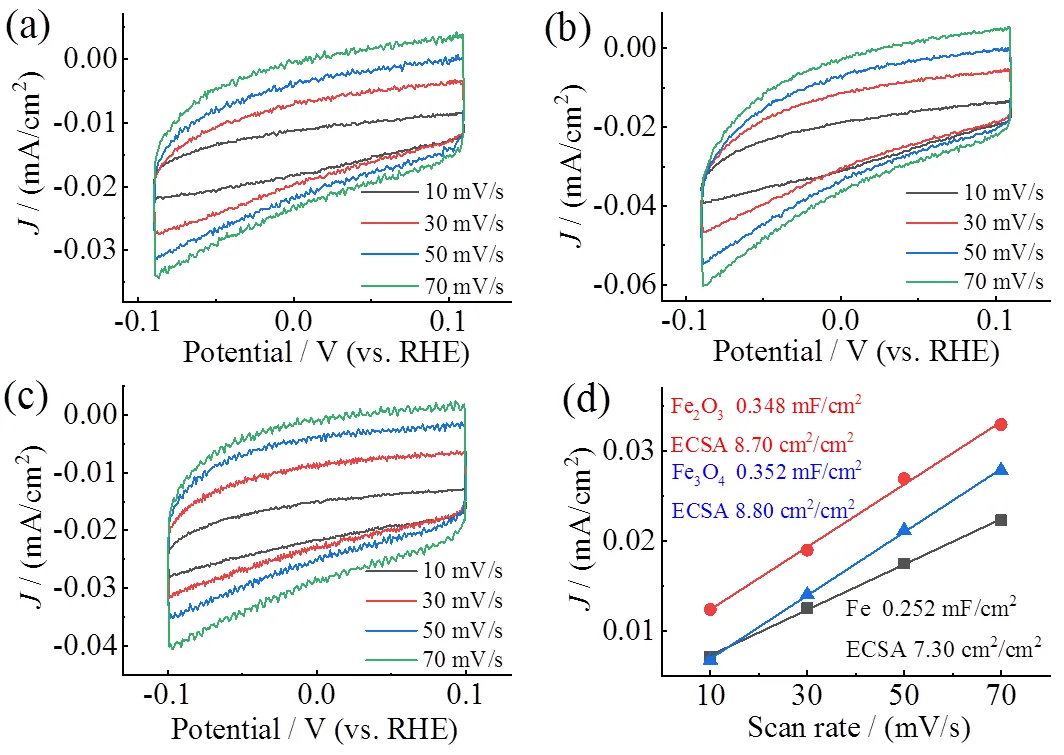
图10 电化学活性面积:Fe(a)、Fe2O3(b)、Fe3O4(c)在10 ~ 70 mV/s的CV曲线;(d)电流密度和扫描速度的关系
2.3.3 催化剂的稳定性
测试了三种铁系材料电催化性能的循环性和稳定性。考虑到−0.83 V为Fe和Fe3O4的最佳NARR电压(FENH3最大),因此选取电解电压为−0.83 V,连续电解6 h后更新电解液,并检测每次循环结束后电解液中产出的NH3(图11a ~ 图11c)。结果表明,即使长时间电解,三种材料在整个电解过程仍可保持着较高水平的FENH3,特别是Fe2O3(图11b),在电解6 h后对应的FENH3可达80.8%,并在往后的三次循环中均保持在81%以上。持续工作24 h后,FENH3无衰减迹象,说明了Fe2O3具有十分出色的催化稳定性和可循环性。
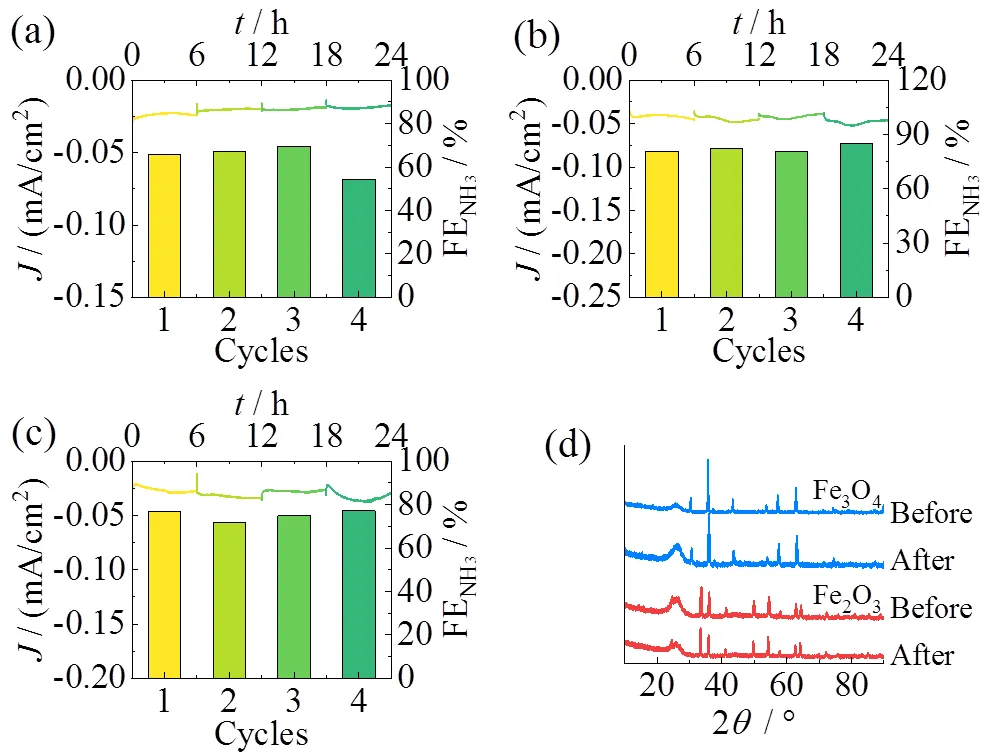
图11 Fe(a)、Fe2O3(b)和Fe3O4(c)在−0.83 V电压下J-t曲线以及每次循环的FENH3;(d)Fe3O4和Fe2O3在反应前后XRD对比
另外,考虑到NARR过程所施加的还原电势极可能使催化剂中高价态的铁离子发生还原反应,因此测试并对比了电解前后的Fe3O4和Fe2O3电极的XRD谱图(图11d)。结果显示,Fe3O4和Fe2O3的衍射图谱均无明显改变,未观测到新物种的形成,表明其在催化过程中的稳定性。
2.4 技术经济分析
基于当前风电、光伏发电、核电三种新能源发电技术下的电价水平,计算出了还原NO3−所获得每吨NH3所需的用电成本(假定FENH3= 100%),并以此进行初步的技术经济分析(由于本分析目的在于初步评估NARR的研究价值,因此成本计算只涉及初步的物料转化,其他基础投入和人工维护在此未做考虑)。另外,考虑到NO3−转化率为50%时对应的产品为NH4NO3,具有广阔的市场价值,因此也考虑了该路线的经济性。
图12所示,水平线分别为目前NH3(实线,¥4 800元/t)和NH4NO3(虚线,¥4 000元/t)的国内市场价格;绿色、蓝色和黄色斜线分别代表基于目前国内风电[¥0.35元/(kW∙h)]、光伏发电[¥0.48元/(kW∙h)]和核电[¥0.36元/(kW∙h)]的价格水平下,产出每吨NH3(实线)和NH4NO3(虚线)的用电成本和电解电压的函数关系[39-40]。从图中可以看出,若采用产NH3路线,当采用风电作为生产电源并实现在1.09 V的电压开展生产时,即可实现用电成本与产出产品价格持平;继续降低电压即可产生利润。若目标产品为NH4NO3,则由于硝酸转化率降低了50%,每吨产品的生产成本将进一步减少,只需使用4.27 V以下的风电便可开始产生经济效益。相比而言,NH4NO3生产路线的成本更低,经济性更好。考虑到当前的合成氨工业高度依赖化石燃料,生产过程会产生大量二氧化碳,随着“双碳”目标的提出,各类高碳强度行业,包括传统的脱硝技术,相关生产成本必将随着政策的收紧而越来越高。而电催化NARR过程可利用可再生电能进行氨的生产,有望实现零碳排放,高度契合国家的战略发展需求;同时,该技术路线还能对接污水处理行业,消纳大量的含硝酸盐污水,经济效益和社会效益也将进一步凸显。考虑到随着可再生发电行业规模的持续扩大,未来NARR的生产用电成本也有望进一步降低。以铁系材料电催化NARR技术路线将具有非常广阔的发展前景。
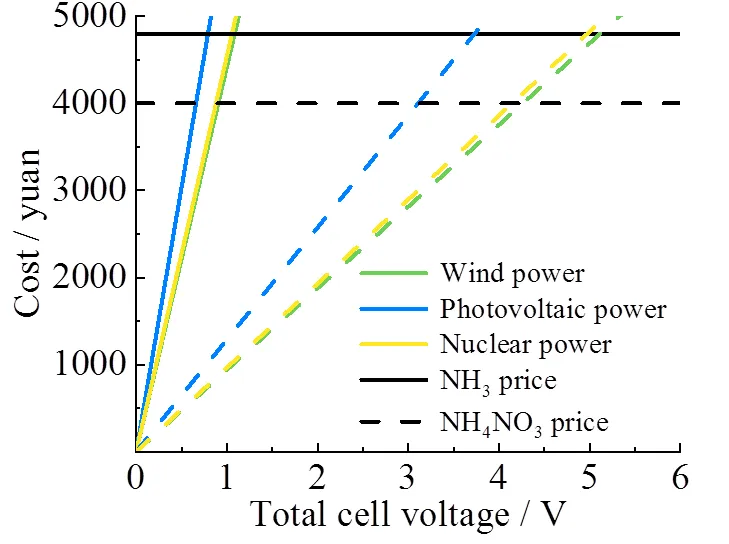
图12 基于新能源的电催化硝酸盐还原产品的经济性分析
3 结 论
以单质Fe、Fe2O3和Fe3O4作为电催化硝酸盐还原合成氨反应的电催化剂,验证了铁系催化剂的电催化活性,而且通过对比发现Fe2O3的催化性能十分优异,其生成氨的法拉第效率可达到88%、氨电流密度达到43.1 mA/cm2,将其作为催化材料具有十分巨大的发展潜力。另外,从用电成本和目标产品的角度进行分析,系统论证了结合可再生电能进行NARR生产的经济可行性。以铁系催化剂进行新能源电催化硝酸盐还原的新型合成氨技术路线契合国家“双碳”目标发展战略,在未来具有非常广阔的发展前景和经济效益。
[1] ROSCA V, DUCA M, DE GROOT M T, et al. Nitrogen cycle electrocatalysis[J]. Chemical reviews, 2009, 109(6): 2209-2244. DOI: 10.1021/cr8003696.
[2] ERISMAN J W, SUTTON M A, GALLOWAY J, et al. How a century of ammonia synthesis changed the world[J]. Nature geoscience, 2008, 1(10): 636-639. DOI: 10.1038/ngeo325.
[3] HIRAKAWA H, HASHIMOTO M, SHIRAISHI Y, et al. Selective nitrate-to-ammonia transformation on surface defects of titanium dioxide photocatalysts[J]. ACS catalysis, 2017, 7(5): 3713-3720. DOI: 10.1021/acscatal.7b00611.
[4] KLERKE A, CHRISTENSEN C H, NØRSKOV J K, et al. Ammonia for hydrogen storage: challenges and opportunities[J]. Journal of materials chemistry, 2008, 18(20): 2304-2310. DOI: 10.1039/b720020j.
[5] GUO J P, CHANG F, WANG P K, et al. Highly active MnN-Li2NH composite catalyst for producing CO-free hydrogen[J]. ACS catalysis, 2015, 5(5): 2708-2713. DOI: 10.1021/acscatal.5b00278.
[6] KOBAYASHI H, HAYAKAWA A, SOMARATHNE K D K A, et al. Science and technology of ammonia combustion[J]. Proceedings of the combustion institute, 2019, 37(1): 109-133. DOI: 10.1016/j.proci.2018.09.029.
[7] MILTON R D, ABDELLAOUI S, KHADKA N, et al. Nitrogenase bioelectrocatalysis: heterogeneous ammonia and hydrogen production by MoFe protein[J]. Energy & environmental science, 2016, 9(8): 2550-2554. DOI: 10.1039/c6ee01432a.
[8] GONG Y T, WU J Z, KITANO M, et al. Ternary intermetallic LaCoSi as a catalyst for N2activation[J]. Nature catalysis, 2018, 1(3): 178-185. DOI: 10.1038/s41929-017-0022-0.
[9] CHEN J G, CROOKS R M, SEEFELDT L C, et al. Beyond fossil fuel-driven nitrogen transformations[J]. Science, 2018, 360(6391): eaar6611. DOI: 10.1126/science. aar6611.
[10] WOOD S W, COWIE A. A review of greenhouse gas emission factors for fertiliser production[Z]. The Climate Technology Centre and Network (CTCN): Copenhagen, Denmark, 2004.
[11] HE D P, LI Y M, OOKA H, et al. Selective electrocatalytic reduction of nitrite to dinitrogen based on decoupled proton-electron transfer[J]. Journal of the american chemical society, 2018, 140(6): 2012-2015. DOI: 10.1021/jacs.7b12774.
[12] SHEN B X, LIU Z, XU H, et al. Enhancing the absorption of elemental mercury using hydrogen peroxide modified bamboo carbons[J]. Fuel, 2019, 235: 878-885. DOI: 10.1016/j.fuel.2018.08.082.
[13] LI J, ZHAN G M, YANG J H, et al. Efficient ammonia electrosynthesis from nitrate on strained ruthenium nanoclusters[J]. Journal of the american chemical society, 2020, 142(15): 7036-7046. DOI: 10.1021/jacs.0c00418.
[14] GARCIA-SEGURA S, LANZARINI-LOPES M, HRISTOVSKI K, et al. Electrocatalytic reduction of nitrate: fundamentals to full-scale water treatment applications[J]. Applied catalysis B: environmental, 2018, 236: 546-568. DOI: 10.1016/j.apcatb.2018.05.041.
[15] MCENANEY J M, BLAIR S J, NIELANDER A C, et al. Electrolyte engineering for efficient electrochemical nitrate reduction to ammonia on a titanium electrode[J]. ACS sustainable chemistry & engineering, 2020, 8(7): 2672-2681. DOI: 10.1021/acssuschemeng.9b05983.
[16] VAN DERHOEK J P, KLAPWIJK A. Nitrate removal from ground water[J]. Water research, 1987, 21(8): 989-997.DOI:10.1016/S0043-1354(87)80018-0.
[17] MA X J, LI M, FENG C P, et al. Electrochemical nitrate removal with simultaneous magnesium recovery from a mimicked RO brine assisted bychloride ions[J]. Journal of hazardous materials, 2020, 388: 122085. DOI: 10.1016/j.jhazmat.2020.122085.
[18] XU S, ASHLEY D C, KWON HY, et al. A flexible, redox-active macrocycle enables the electrocatalytic reduction of nitrate to ammonia by a cobalt complex[J]. Chemical science, 2018, 9(22): 4950-4958. DOI: 10.1039/c8sc00721g.
[19] PENNINO M J, COMPTON J E, LEIBOWITZ S G. Trends in drinking water nitrate violations across the United States[J]. Environmental science & technology, 2017, 51(22): 13450-13460. DOI: 10.1021/acs.est.7b04269.
[20] EPSZTEIN R, NIR O, LAHAV O, et al. Selective nitrate removal from groundwater using a hybrid nanofiltration-reverse osmosis filtration scheme[J]. Chemical engineeringjournal, 2015, 279: 372-378. DOI: 10.1016/j.cej.2015.05.010.
[21] ALIKHANI M, MOGHBELI M R. Ion-exchange polyHIPE type membrane for removing nitrate ions: preparation, characterization, kinetics and adsorption studies[J]. Chemical engineering journal, 2014, 239: 93-104. DOI: 10.1016/j.cej.2013.11.013.
[22] SAMATYA S, KABAY N, YUKSELÜ, et al. Removal of nitrate from aqueous solution by nitrate selective ion exchange resins[J]. Reactive and functional polymers, 2006, 66(11): 1206-1214. DOI: 10.1016/j.reactfunctpolym.2006.03.009.
[23] GHAFARI S, HASAN M, AROUA M K. Bio-electrochemical removal of nitrate from water and wastewater - A review[J]. Bioresource technology, 2008, 99(10): 3965-3974. DOI: 10.1016/j.biortech.2007.05.026.
[24] WEI X, VOGEL D, KELLER L, et al. Microtubular gas diffusion electrode based on ruthenium-carbon nanotubes for ambient electrochemical nitrogen reduction to ammonia[J]. ChemElectroChem, 2020, 7(22): 4679-4684. DOI: 10.1002/celc.202001370.
[25] SONG X S, LI Q, YAN D H. Nutrient removal by hybrid subsurface flow constructed wetlands for high concentrationammonia nitrogen wastewater[J]. Procedia environmental sciences, 2010, 2: 1461-1468. DOI:10.1016/j.proenv. 2010.10.159.
[26] XU Y T, PENG Z G, HAN Y, et al. Insight into hydrogenation selectivity of the electrocatalytic nitrate-to-ammonia reduction reaction via enhancing the proton transport[J]. ChemSusChem, 2022, 15(6): e202102450. DOI: 10.1002/cssc.202102450.
[27] XU H, MA Y Y, CHEN J, et al. Electrocatalytic reduction of nitrate - a step towards a sustainable nitrogen cycle[J]. Chemical society reviews, 2022, 51(7): 2710-2758. DOI: 10.1039/d1cs00857a.
[28] LIU M, MAO Q Q, SHI K K, et al. Electroreduction of nitrate to ammonia on palladium-cobalt-oxygen nanowire arrays[J]. ACS applied materials & interfaces, 2022, 14(11): 13169-13176. DOI: 10.1021/acsami.1c19412.
[29] LEI X H, LIU F, LI M, et al. Fabrication and characterization of a Cu-Pd-TNPs polymetallic nanoelectrodefor electrochemically removing nitrate from groundwater[J]. Chemosphere, 2018, 212: 237-244. DOI: 10.1016/j.chemosphere.2018.08.082.
[30] BADEA G E. Electrocatalytic reduction of nitrate on copper electrode in alkaline solution[J]. Electrochimica acta, 2009, 54(3): 996-1001. DOI: 10.1016/j.electacta.2008.08.003.
[31] 张承帅, 李莉, 张长青. 中国铁资源利用现状述评[C]//第一届全国青年地质大会论文集. 福州: 中国地质学会, 2013.
[32] WANG Y T, ZHOU W, JIA R R, et al. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia[J]. Angewandte chemieinternational edition, 2020, 59(13): 5350-5354. DOI: 10.1002/anie.201915992.
[33] LI H L, CHAMBERS J Q, HOBBS D T. Electroreduction of nitrate ions in concentrated sodiumhydroxide solutions at lead, zinc, nickel and phthalocyanine-modified electrodes[J]. Journal of applied electrochemistry, 1988, 18(3): 454-458. DOI: 10.1007/bf01093762.
[34] URETA-ZAÑARTUS, YÁÑEZ C. Electroreduction of nitrate ion on Pt, Ir and on 70:30 Pt: Ir alloy[J]. Electrochimica acta, 1997, 42(11): 1725-1731. DOI: 10.1016/s0013-4686(96)00372-6.
[35] WHITE R E. Modern Aspects of Electrochemistry 45[M].New York: Springer, 2009. 1-61. DOI: 10.1007/978-1-4419-0655-7.
[36] WANG Z X, RICHARDS D, SINGH N. Recent discoveries in the reaction mechanism of heterogeneous electrocatalytic nitrate reduction[J]. Catalysis science & technology, 2021, 11(3): 705-725. DOI: 10.1039/d0cy02025g.
[37] DE GROOT M T, KOPER M T M. The influence of nitrate concentration and acidity on the electrocatalytic reduction of nitrate on platinum[J]. Journal of electroanalytical chemistry, 2004, 562(1): 81-94. DOI: 10.1016/j.jelechem.2003.08.011.
[38] LI P P, JIN Z Y, FANG Z W, et al. A single-site iron catalyst with preoccupied active centers that achieves selective ammonia electrosynthesis from nitrate[J]. Energy & environmental science, 2021, 14(6):3522-3531. DOI:10.1039/D1EE00545F.
[39] 张运洲, 黄碧斌. 中国新能源发展成本分析和政策建议[J]. 中国电力, 2018, 51(1): 10-15. DOI:10.11930/j. issn.1004-9649.201711268.
[40] 王宇, 朱沈超, 陈芳斌, 等. 中国核电与可再生能源发电协调发展初探[J].可再生能源, 2021, 39(8): 1069-1077.DOI:10.3969/j.issn.1671-5292.2021.08.013.
Iron-Based Inorganic Materials for New Energy-Based Electrocatalytic Nitrate-to-Ammonia Reduction Reaction
REN Ke-cheng1,2,3, XU Yan-tong2,3, CAO Yan1,2,3
(1. School of Chemistry and Chemical Engineering, Anhui University, Hefei 230601, China;2. Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou 510640, China;3. Guangdong Provincial Key Laboratory of New and Renewable Energy Research and Development, Guangzhou 510640, China)
Nitrate is a kind of widespread environmental pollutants, while its corresponding reduction state, ammonia, is an important chemical raw material and agricultural fertilizer. Therefore, exploring the direct chemical transformation between nitrate and ammonia is of great significance technically and economically, especially for the technological upgrade of the energy-intensive ammonia industry driven by the carbon neutrality target and the possibility of ammonia in being the next generation of hydrogen carrying fuel. Among them, the electrocatalytic technology can combine the nitrate-to-ammonia reduction reaction (NARR) process with renewable electricity, building up a new recycling technology and economic system based on the green and low-carbon artificial “nitrogen cycle” of nitrogen-containing chemicals. Therefore, electrocatalytic NARR is an effective route to resolve the problems of high CO2emission and dependence on fossil energy of ammonia industry. Iron-based inorganic materials have been widely applied in the traditional ammonia industry, which also has a huge cost advantage over other materials. In this paper, metallic Fe, Fe2O3and Fe3O4were employed as catalytic materials to explore the optimal chemical state of iron-based catalysts on electrocatalytic NARR. Typically, in the potential range between −0.53 V to −0.93 V (vs. reversible hydrogen electrode, RHE), Fe2O3exhibited the most excellent catalytic activity, whose Faradaic efficiency toward NH3(FENH3) could reach up to 88%. Moreover, the corresponding partial current density of NH3(NH3) was 43.1 mA/cm2, and the NH3formation rate (NH3) was 0.20 mmol/(cm2·h). In addition, technical routes toward NH3and NH4NO3products could be switched via controlling nitrate conversion ratio. Comparing the corresponding electricity cost and product market price, it was verified that both routes (especially the NH4NO3route), were economically feasible and the iron-based inorganic materials have a very huge market potential in electrocatalytic NARR.
iron-based materials; nitrate reduction; ammonia synthesis; electrocatalysis
2095-560X(2022)03-0209-09
TK01+9
A
10.3969/j.issn.2095-560X.2022.03.004
2022-03-01
2022-04-04
国家自然科学基金项目(22178339);广州市科技计划项目(202102020866);中国科学院特别研究助理资助项目(1190000058);中国博士后科学基金项目(2021M690151);广东省基础与应用基础研究基金项目(21201910240004684);广东省新能源和可再生能源研究开发与应用重点实验室项目(2021000037)
曹 晏,E-mail:caoyan@ms.giec.ac.cn
# 该作者对论文有同等贡献
任柯丞(1996-),男,硕士研究生,主要从事电化学合成氨催化转化研究。
许彦桐(1992-),男,博士,主要从事电化学驱动的能量物质循环过程与相关功能材料的分子设计研究。
曹 晏(1968-),男,博士,研究员,以化学反应与分离工程学科为主线,交叉融合材料化学、电化学、分析化学和量子化学等学科,从事能源转化、环境保护及废弃物资源回收等方向的基础研究与技术研发。
猜你喜欢
电池(2022年2期)2022-11-07
电池(2022年4期)2022-11-07
读书文摘(下半月)(2021年1期)2021-06-28
学校教育研究(2020年4期)2020-04-10
中国校外教育(中旬)(2018年9期)2018-09-30
科学与财富(2017年32期)2017-12-20
中小企业管理与科技·下旬刊(2017年8期)2017-09-13
农民科技培训(2009年1期)2009-02-17
中学生数理化·高一版(2008年4期)2008-11-15
中学生数理化·高二版(2008年6期)2008-11-12
