
Mg-doped layered oxide cathode for Na-ion batteries
2022-06-29 08:56:40YuejunDing丁月君FeixiangDing丁飞翔XiaohuiRong容晓晖YaxiangLu陆雅翔andYongShengHu胡勇胜
Chinese Physics B 2022年6期
关键词:飞翔
Yuejun Ding(丁月君) Feixiang Ding(丁飞翔) Xiaohui Rong(容晓晖)Yaxiang Lu(陆雅翔) and Yong-Sheng Hu(胡勇胜)
1Key Laboratory for Renewable Energy,Beijing Key Laboratory for New Energy Materials and Devices,Beijing National Laboratory for Condensed Matter Physics,Institute of Physics,Chinese Academy of Sciences,Beijing 100190,China
2Center of Materials Science and Optoelectronics Engineering University of Chinese Academy of Sciences,Beijing 100190,China
Keywords: copper redox,layered oxide,cathode,Na-ion batteries,energy storage
1. Introduction
Li-ion batteries (LIBs) are the optimum choice for portable electronic devices and electric vehicles due to the high energy density and energy conversion efficiency. However,the energy density is not the primary consideration when it comes to large-scale or stationary electrical energy storage systems. Instead,Na-ion batteries(NIBs)as a helpful complement have drawn increasing interest owing to the abundance and low cost of sodium resources, as well as the similar Na+storage mechanism to that of the LIBs.[1,2]Extensive efforts have been made and improve the comprehensive performance of NIBs.[3–5]It is of great importance for the improvement of the performance of NIBs to explore electrode materials with high capacity, high voltage, and excellent cycle stability.[6,7]Layered transition metal(TM)oxides(NaxTMO2)are an important type of cathode materials due to their easy synthesis,remarkable Na+intercalation/deintercalation reversibility,excellent diffusion kinetics, and high theoretical capacity.[8–10]Driven by the urgent demand for electrode materials with high energy density,long-cycling stability and low-cost in the commercial application of NIBs,many cathode materials with superior electrochemical performance have been designed and developed in recent years.[6,11,12]
Cu2+was firstly found to be electrochemically active in P2-Na0.68Cu0.34Mn0.66O2compound.[13]Compared with other reported redox couple of +2/+3 valence in layered oxides, such as Ni3+/Ni2+, the advantage of Cu3+/Cu2+redox couple lies in its higher reaction potential and relatively low-cost. Subsequently, a series of Cu-containing layered oxides have been developed.[14–31]It was found that the Cu2+substitution not only contributes to charge compensation,[14,15,17,20,21]but also improves cycling performance via suppressing phase transition,[22,23,25,26]or mitigating TM dissolution,[27,28]which enhance Na+diffusion kinetics and promote the rate performance of the cathode materials.[29–31]However, among the Cu-contained materials reported above, only part of the Cu3+/Cu2+redox couple participated in the redox reaction, and the activity of Cu3+/Cu2+redox for this type of materials needs to be further improved. The previous studies showed that Mg2+substitution generally contributes to stabilizing the crystal structure. For example, Na[Ni4/9Mn1/3Mg1/18Ti1/6]O2goes through more continuous phase transitions and lattice parameter variation than the undoped one;[32]Mg dopant in O3-NaMg0.05[Ni1/3Fe1/3Mn1/3]0.95O2suppresses the occurrence of irreversible phase transitions as well as structural degradation;[33]Mg doped P3-Na2/3Ni1/4Mg1/12Mn2/3O2is stable against water and air.[34]However, to date, there is yet to be a systematic study on the effect of Mg-doping on the electronic structure of the layered oxides cathode.
Compared with the P2-type layered oxides, the O3-type layered oxides have a considerably higher initial interlayer Na+content and thus can provide more reversible Na+de-intercalation and higher initial Coulombic efficiency. This is crucial for improving the reversible capacity of the Na-ion full-cell.[35]Herein, we report a novel Mg-doped O3-type Cu–Fe–Mn based layered oxide,O3-Na0.90Mg0.08Cu0.22Fe0.30Mn0.40O2(abbreviated as NMCFM), with more reversible intercalation/deintercalation of Na+compared with undoped O3-Na0.90Cu0.22Fe0.30Mn0.48O2(abbreviated as NCFM).[36]This work reveals, for the first time, that Mg2+doping could effectively promote the electrochemistry of the Cu3+/Cu2+redox,thus increasing the discharge voltage in O3-type materials. Based on the analysis ofin situx-ray diffraction (XRD) result, we found that the irreversible phase transition occurring at higher-voltage gives rise to the significantly increased polarization and capacity fading.By reducing the charge cutoff voltage to 3.85 V,we found that NMCFM still delivered a reversible capacity of 100 mAh/g with long-cycling stability.
2. Experimental details
2.1. Materials synthesis
The O3-Na0.90Mg0.08Cu0.22Fe0.30Mn0.40O2and the referential sample O3-Na0.90Cu0.22Fe0.30Mn0.48O2were synthesized by a simple solid-state reaction at 850°C in air atmosphere using precursors of Na2CO3(Alfa, 99.5%), MgO(Aladdin, 99.9%), CuO (Aladdin, 99.9%), Fe2O3(Aladdin,99.8%),and MnO2(Aladdin,99.5%).
2.2. Structure and morphology characterization
The XRD patterns were collected on a Bruker-AXS D8 Advance instrument using CuKαradiation. The structural refinement was performed by Rietveld refinements using the Fullprof suite. The special cell forin situXRD experiment during the first charge and discharge was assembled with the Al foil as the x-ray window and current collector. The actual chemical composition of the final materials was determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES, Shimadzu, ICPS-8100). Scanning electron microscopy (SEM) measurements were carried out on a Hitachi S-4800(10 kV).
2.3. Electrochemical characterization
The working cathodes were fabricated by mixing active material with carbon nanotube and polytetrafluoroethylene(in a weight ratio of 8:1:1),which were rolled into thin films with the loading mass of the active material of 4–7 mg/cm2. The prepared electrodes were dried at 100°C under vacuum for 12 h and then were fabricated into CR2032 coin-type cells in an argon-filled glove box(H2O,O2<0.1 ppm). Commercial glass fibers and sodium metal were employed as the separator and counter/reference electrode,respectively. 1 M NaClO4in propylene carbonate/ethylene carbonate/dimethyl carbonate(PC/EC/DMC=1:1:1 in volume) with fluoroethylene carbonate (FEC, 2% in volume) was used as the electrolyte. The galvanostatic charge and discharge measurements were carried out on a Neware CT4000 battery test system in various voltage ranges and current rates under room temperature,and a rate of 1 C corresponding to 100 mA/g. The CV tests were performed on an electrochemical workstation CHI 760e in the voltage range of 2.4–3.9 V or 2.4–4.5 V vs.Na+/Na at various scan rates. GITT was measured by applying the repeated current pulses for 30 min at a current density of 15 mA/g followed by relaxation for 2 h. The electrochemical impedance spectroscopy(EIS)of the coin cells was recorded by electrochemical workstation CHI 760e in the discharged state of 2.4 V at room temperature between 1 MHz and 10 mHz with an AC voltage of 5 mV.
3. Results and discussion
The Mg2+doped NMCFM could be synthesized by means of solid-state reaction at 850°C in air atmosphere. As shown in Fig.1(a),NMCFM was characterized as a typicalα-NaFeO2structure,belonging to theR-3mspace group,similar to NCFM.A few weak diffraction peaks in XRD spectrum of NMCFM corresponded to CuO impurities. According to the XRD refinement result, part of the Mn3+with a smaller ion radius was replaced by Mg2+with a larger ion radius(0.62 °A vs. 0.72 °A). The lattice parametera=2.96877 °A increased whilec=16.20971 °A decreased comparing with the lattice parameter of NCFM(a=2.9587 °A,c=16.3742 °A).The ICP results confirmed that stoichiometric ratio of various elements was nearly the same as the designed one. Moreover,the chemical valence state of Mn in the material could be calculated to be +4 based on the electroneutrality principle. Evidently,the design objective material with free of Mn3+was achieved,demonstrating that the Mg2+was a good alternative to replace Mn3+.
Furthermore, the size and morphology of NMCFM particles were studied by SEM, showing a particle size ranged from 1 μm to 3 μm. Meanwhile,almost all the particles were polyhedral and plate-like with smooth surfaces. Based on the energy dispersive spectroscopy(EDS)results(Fig.1(b)), Cu,Fe, Mn and Mg elements were uniformly distributed in the material bulk, indicating that Mg ions were uniformly doped into NMCFM.
Galvanostatic charge–discharge test was carried out on NMCFM at a current rate of 0.2 C in Na-ion half-cells.The first-cycles of charging and discharging curves at different voltage intervals were compared, as shown in Fig. 2(a),showing that the initial Coulombic efficiency decreases as the charge cut-off voltage increases. In addition, the polarization in a narrow voltage range is significantly reduced in comparison to that in a wider voltage window. It is possibly related to the irreversible migration of Fe4+into the Na+layer upon increasing Fe3+/Fe4+redox reaction,which deteriorates the Na+diffusion dynamics.[20,37]Besides,when the charging voltage was above 4.0 V,a second voltage platform appeared,indicating another phase transformation occurred in the highvoltage range after the O3-P3 two-phase reaction. In consequence,both the reactions mentioned above may directly lead to the increase of polarization.

Cyclic voltammetry (CV) was employed to compare the kinetics property of these two oxide electrodes. As shown in Fig. 2(b), both oxide cathodes were tested with the potential ranges of 2.4–3.9 V using scan rates of 0.08 mV/s, demonstrating good reversibility in the first two cycles. A pair of reversible redox peaks(at 3.203 V and 3.014 V)are shown in the cyclic voltammogram of NCMFM, and a similar pair of reversible redox peaks(at 3.160 V and 2.941 V)are also displayed for NCFM.The redox peak potentials of NMCFM are higher than those of NCFM,indicating that Mg doping could increase the reaction potential of the oxide cathode. Furthermore, the electrochemical polarization (0.189 V) calculated between the redox peaks of NMCFM is also smaller than that of NCFM(0.219 V),indicating that Mg doping could enhance the phase stability as well.
It can be seen from Figs. 2(c) and 2(d), NMCFM delivers a reversible capacity of 107 mAh/g in the voltage range of 2.4–3.9 V, corresponding to the reversible intercalation/deintercalation of 0.43 Na+,with high initial Coulombic efficiency of 92%. Whereas within the same voltage interval, NCFM only delivers a reversible capacity of 80 mAh/g,corresponding to the reversible intercalation/deintercalation of 0.32 Na+. Similarly, in the voltage range of 2.4–4.0 V,NMCFM and NCFM respectively deliver a reversible capacity of 117.7 mAh/g and 90 mAh/g, corresponding to the reversible intercalation/deintercalation of 0.47 and 0.37 Na+ions. Therefore, compared to NCFM, additional 0.10 Na+ions are available for NMCFM, resulting in the specific energy of 347.9 Wh/kg and 262.6 Wh/kg in the voltage range of 2.4–3.9 V,as well as 394.4 Wh/kg and 295.9 Wh/kg in the voltage range of 2.4–4.0 V for NMCFM and NCFM, respectively.As shown in Fig.2(e),the NMCFM cathode can deliver a highly reversible capacity of~100 mAh/g in the voltage range of 2.4–3.85 V.As for the rate performances in the voltage range of 2.4–3.9 V, the reversible capacities of NMCFM are 107.3 mAh/g, 101.4 mAh/g, 93.3 mAh/g, 86.1 mAh/g,79 mAh/g and 65 mAh/g at 0.1 C,0.2 C,0.5 C,1 C,2 C and 5 C(Fig.3(a)),respectively. In comparison,the reversible capacities of NCFM are 88.1 mAh/g,83.6 mAh/g,77.3 mAh/g,74.2 mAh/g,71 mAh/g and 64.1 mAh/g at 0.1 C,0.2 C,0.5 C,1 C,2 C and 5 C(Fig.3(b)),respectively. As a result,it could be concluded that Mg2+doping contributes to the improvement of the electrochemical activity of Cu3+/Cu2+redox.Furthermore,as shown in Figs.3(c)–3(d),NMCFM exhibited outstanding cyclability with capacity retention of 88% after 200 cycles at 0.2 C in 2.4–3.85 V; even at the higher rate of 1 C,75%capacity was still retained after 500 cycles.[11]

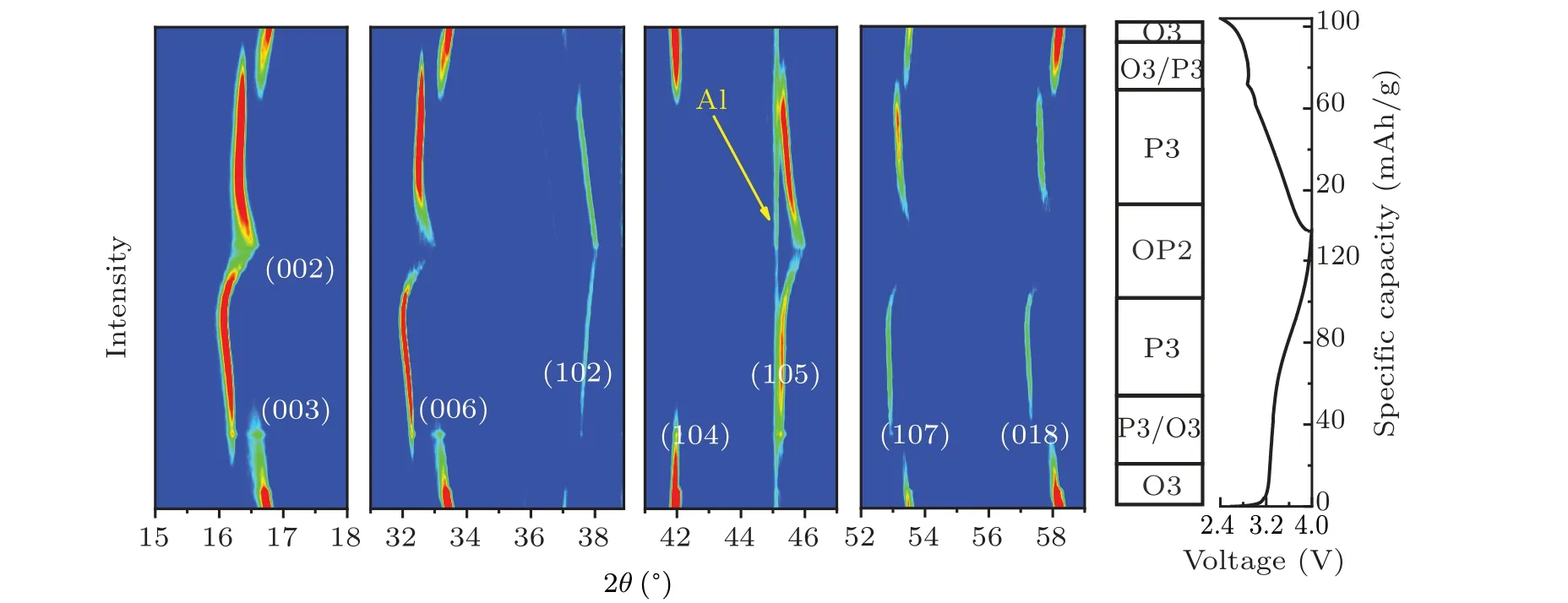
In situx-ray diffraction characterization was performed in order to capture the structural evolution process of NMCFM during the Na+insertion and extraction process. The correspondingin situXRD spectra were collected during the initial charge and discharge process at the rate of 0.1 C in the voltage range of 2.4–4.0 V and are shown in Fig. 4. When Na+extraction amountxwas lower than 0.1,or between 0.22 and 0.45,the material maintained O3 and P3 phase structures,respectively. Meanwhile, the diffraction peak(00l)shifted to a lower angle as the charging voltage increased. Evidently,along with the content of interlayered Na+decreased,the electrostatic repulsion between TMO2layers would increase,leading to an increase in the interlayer spacing along thec-axis.The single-phase solid solution reaction was consistent with electrochemical behavior in the corresponding interval of voltage. When the Na+extraction amountxwas in the range from 0.12 to 0.22, the intensity of (003), (006) and (104) diffraction peaks that belonged to O3 phase declined gradually; in the meantime, the (003), (006) and (105) diffraction peaks that belonged to P3 phase started to appear,which implies O3 phase and P3 phase coexist in this process.[38]When the deintercalated Na+ion amount exceeded 0.45,the(00l)diffraction peaks moved towards a higher angle.[38]This might be due to the crystal structure starting to shrink upon the formation of more Na+vacancies. Furthermore,the(002)diffraction peak belonging to a new phase was found,which was identified as the OP2-like phase.[32]What is more, the intensities of various diffraction peaks dramatically decreased with peak-width broadening,which is due to a larger cubic deformation of OP2-like phase. Moreover, either the lattice distortion or the irreversible migration of Fe4+to the Na+layer could hinder the Na+diffusion,which is consistent with the finding of the significant decline ofDNa+calculated by GITT.In short,during the initial charge process,the layered oxide cathode underwent the phase transformation of O3→P3→OP2-like, meanwhile the discharge process displayed an opposite phase transformation. However, when it was discharged to 2.4 V, the (003)diffraction peak angle of reformed O3 phase was lower than that of the pristine sample,which implies the Na+ion content reduced slightly after the initial insertion and extraction cycle.
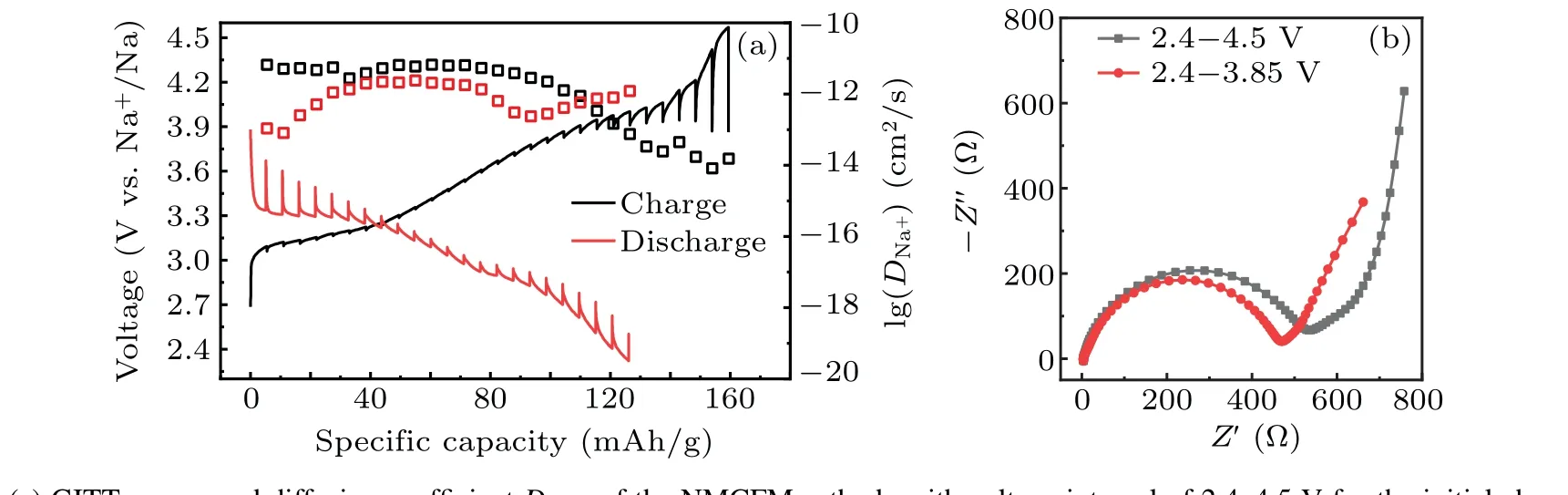
GITT is a common electrochemical technique that is used for analyzing the diffusion ability of Na+in electrode materials. As can be seen in Fig. 5(a), the apparent diffusion coefficientDNa+is little changed in the voltage range of 2.4–3.9 V, reasonably maintaining at the 10-12cm2/s order of magnitudes.[39]However,DNa+declines sharply in the voltage window ranged from 3.9 V to 4.5 V,which decreases to below 10-13cm2/s especially when it is charged to above 4.15 V.The reason is that Na+diffusion is severely hampered due to the generation of a new phase.[40]Furthermore,EIS of the fabricated coin cells consisting of NMCFM electrodes was measured and analyzed after the first cycle in the voltage ranges of 2.4–3.85 V and 2.4–4.5 V,respectively. It is obvious that the electrode cycled in the voltage range of 2.4–3.85 V exhibits lower resistance than the electrode cycled in the voltage range of 2.4–4.5 V,suggesting the reduced Na+diffusion resistance and charge-transfer resistance when cycled in narrow voltage range. These results suggest that Mg2+doping stabilizes the layered structures in a suitable voltage range, thus enhancing the electrochemical properties of the layered oxides. Our findings from the systematic studies on the influence of doping metal are instructive and will stimulate further research for enhancing the structural stability and electrochemical performance of the O3-type layered oxide cathode at higher voltages.
We also assembled full cells with hard carbon (HC) as the anode to demonstrate the excellent electrochemical performance of the NMCFM cathode. Figures 6(a) and 6(b) show that a reversible storage capacity of~92 mAh/g(based on the mass of cathode active material in the cell)was achieved in the voltage range of 0.5–3.85 V at 0.1 C.The specific energy density was calculated to be 262 Wh/kg based on the total mass of cathode and anode active materials. The reversible capacities were 70 mAh/g and 47 mAh/g at the rates of 2 C and 5 C(Fig.6(a)),respectively. The cell also presented excellent cycling stability and the capacity retention was 96% after 80 cycles at 1 C rate. Thus,the high energy density and cycle stability revealed in the full cells stand a good chance to meet the practical applications and commercial demands henceforward.

4. Conclusions
In summary, we reported an O3-type Mg2+-doped NMCFM (Na0.90Mg0.08Cu0.22Fe0.30Mn0.40O2) cathode material without Mn3+. The XRD and ICP results indicated that Mg2+is a good alternative to replace Mn3+, which is helpful to increase the reversible specific capacity.Both the SEM and EDS results further illustrated the doped element Mg2+was distributed uniformly. Thein situXRD data demonstrated a reversible O3→P3→OP2-like phase→P3→O3 transformation during the initial charge and discharge process in the voltage range of 2.4–4.0 V.Combined with the GITT analysis,the formation of OP2-like phase at high voltage probably contributes to the reduction ofDNa+, leading to the reversibility of Na+intercalation/deintercalation fading eventually. In the voltage range of 2.4–4.0 V, NMCFM delivered a reversible capacity of 118 mAh/g at 0.2 C,which is significantly higher than that of undoped NCFM.Through limiting the voltage within 2.4–3.85 V,the NMCFM cathode exhibited the capacity retention of 88%after 200 cycles at 0.2 C,75%after 500 cycles at 1 C.As a result,considering the involvement of Cu3+/Cu2+redox couple in the charge/discharge process, it can be concluded that Mg2+doping contributes to improve the electrochemical activity of Cu3+/Cu2+. In full cells, it provides a reversible storage capacity of around~92 mAh/g, achieving a specific energy density of 262 Wh/kg with the capacity retention of 96% after 80 cycles at the rate of 1 C. The NMCFM with high specific capacity and good cycling stability stands a good chance to meet the practical applications and commercial demands henceforward.
Acknowledgements
Project supported by the National Natural Science Foundation of China (Grant Nos. 51725206, 52122214, and 52072403), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA21070500),Youth Innovation Promotion Association of the Chinese Academy of Sciences (Grant No. 2020006), and Beijing Municipal Natural Science Foundation, China (Grant No.2212022).
猜你喜欢
军事文摘(2021年18期)2021-12-02 01:27:52
雪豆月读·低年级(2021年9期)2021-09-13 09:15:17
趣味(语文)(2020年5期)2020-11-16 01:34:52
小学生学习指导(低年级)(2019年12期)2019-12-04 03:39:30
快乐语文(2019年15期)2019-08-27 01:14:22
时代邮刊(2019年16期)2019-07-30 08:02:02
快乐语文(2019年9期)2019-01-11 00:52:17
民族音乐(2018年2期)2018-05-26 03:04:37
草原歌声(2017年3期)2017-04-23 05:13:45
小主人报(2016年5期)2016-02-28 20:48:06
- Chinese Physics B的其它文章
- Switchable terahertz polarization converter based on VO2 metamaterial
- Data-driven parity-time-symmetric vector rogue wave solutions of multi-component nonlinear Schr¨odinger equation
- Neutron activation cross section data library
- Multi-phase field simulation of competitive grain growth for directional solidification
- A novel similarity measure for mining missing links in long-path networks
- Effects of electrical stress on the characteristics and defect behaviors in GaN-based near-ultraviolet light emitting diodes