
Synaptopathy in CHMP2B frontotemporal dementia highlights the synaptic vesicle cycle as a therapeutic target
2022-06-29 08:14MirandaRobbinsEmmaClayton
中国神经再生研究(英文版) 2023年2期
Miranda Robbins, Emma L. Clayton
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are both devastating neurodegenerative conditions. Despite affecting different regions of the nervous system (FTD affecting primarily the frontal and temporal lobes, whilst ALS presents with motor neuron loss), there is significant overlap between these conditions in terms of genetics, pathology, and disease mechanisms, and they are therefore often grouped as a spectrum of symptoms under the heading FTD/ALS (Abramzon et al., 2020). Significantly, there is currently no cure for ALS or FTD. However, recent mechanistic insight points to a novel pathway to target for potential therapeutic intervention.
Synapses are the point of communication between neurons. Chemical synapses are composed of presynaptic and postsynaptic compartments, with intricate trafficking and signaling pathways occurring locally within these compartments which are essential for neuronal communication. Dysfunction of these highly regulated and sophisticated synaptic pathways is known as synaptopathy, and synaptopathy is emerging as a significant contributor to the etiology of multiple neurodegenerative diseases. FTD causative mutation in charged multivesicular body protein 2B (CHMP2B) was recently described as a novel synaptopathy (Clayton et al., 2021). This novel synaptopathy is characterized by defective synaptic vesicle trafficking and retention of the select synaptic vesicle (SV)-associated proteins in aged mice, despite significant neuronal and synaptic loss. In conjunction with several recent publications showing FTD- and ALS-associated genes are involved in SV trafficking pathways, the data show that synaptopathy in the form of dysfunctional SV trafficking is a significant and early event in FTD and ALS, and highlights the potential of the SV cycle as an early and targetable pathway for therapeutic intervention in FTD/ALS.
FTD/ALS associated proteins and the synaptic vesicle cycle:
There is significant overlap in the genetics of FTD and ALS - many genes are known to be causal to both FTD and ALS including C9orf72, TAR DNA-binding protein 43 (TDP-43) and fused in sarcoma (FUS), with multiple other genes related to FTD such as microtubule-associated protein tau, progranulin, and CHMP2B (for a review see Abramzon et al., 2020). Significantly, the convergence of many of these FTD- and ALSassociated gene products at the SV cycle and trafficking pathway in the presynaptic compartment indicates that these processes are a significant contributor to disease mechanisms. The SV cycle involves rounds of calciumdependent exocytosis of SVs containing neurotransmitters into the synaptic cleft, followed by endocytosis of membrane and proteins for the reformation of vesicles necessary for future rounds of neurotransmission (Sudhof, 2004). SVs are arranged in distinct pools in the presynaptic terminal, and vesicle recruitment from these pools depends on the strength and duration of stimulation. Similarly, multiple modes of SV retrieval exist(Figure 1
), which are distinguished by their characteristic molecular mechanisms and the speed of endocytosis (for a comprehensive review on the SV cycle see Chanaday et al., 2019).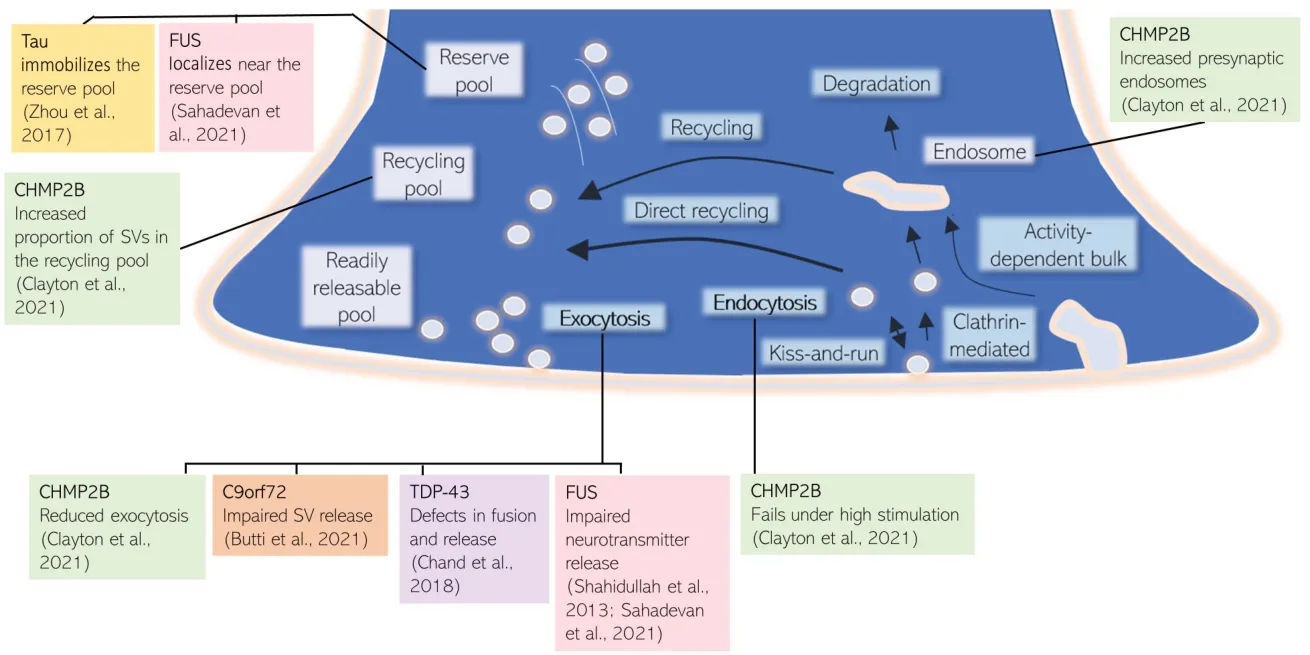
Figure 1|Multiple FTD/ALS associated proteins converge at the synaptic vesicle cycle. CHMP2B, FUS, and Tau have been shown to influence SV pools, whereas CHMP2B, FUS, C9orf72, and TDP-43 can alter SV fusion and release. ALS: Amyotrophic lateral sclerosis; CHMP2B: charged multivesicular body protein 2B; FTD: frontotemporal dementia; FUS: fused in sarcoma; SV: synaptic vesicle; TDP-43: TAR DNA-binding protein 43.
The SV cycling pathway is essential for neurotransmission, and recent studies have highlighted the SV cycle as a convergent pathology in FTD and ALS.
Evidence for the SV cycle as a common pathway affected by FTD and ALS causative mutations: CHMP2B:
CHMP2B is a subunit in the endosomal sorting complex required for transport-III (ESCRT-III) complex. The ESCRT complex has numerous known roles in membrane remodeling, and has recently been shown to have a role in the degradation of synaptic proteins as part of the SV cycle (Sheehan et al., 2016). C-terminal truncation of CHMP2B leads to FTD. This autosomal dominant mutation was initially discovered in one large kindred from Denmark, with a further autosomal dominant CHMP2B mutation found in a Belgian FTLD pedigree (Isaacs et al., 2011). The earliest defects which precede and lead to neuronal loss in mutant CHMP2B FTD are not fully understood. Recently CHMP2B FTD has been reported as a novel synaptopathy, characterized by defective SV trafficking and retention of select SV-associated proteins. In this study, 18-month mutant CHMP2B mice show a surprising retention of certain presynaptic SV-associated proteins, despite significant loss of postsynaptic proteins, indicating synaptic loss concurrent with known neuronal loss. Expression of mutant CHMP2B in primary cortical cultures results in an increased proportion of SVs in the recycling pool, with fewer SVs released during a defined stimulus, and failure of SV endocytosis when the system is highly stimulated. These findings are consistent with the explanation that synaptic vesicle cycling is severely impaired. The model proposed for CHMP2B FTD synaptopathy is through impaired synaptic protein membrane trafficking, leading to reduced exocytosis, altered vesicle pool dynamics, and down-regulated synaptic transmission. In turn, this could cause a loss of synapses and the accumulation of SV proteins in remaining neurons. Although mutant CHMP2B is a minor proportion of FTD cases, insights from this study have highlighted a convergence of pathological disease mechanisms at the presynaptic terminal.C9orf72:
C9orf72 has also been shown to be involved in presynaptic vesicle trafficking. C9orf72 loss-of-function has damaging effects on plasticity including impaired SV release, downregulated synaptic vesicle associated protein 2a, decreased size and number of puncta containing the active zone docking protein Rab3a, a reduced rate of SV cycling, and, ultimately, reduced colocalization of pre- and post-synaptic proteins in a zebrafish model of disease (Butti et al., 2021). C9orf72 mutation has additionally been linked to impaired synaptic function through electrophysiological impairments including enhanced synaptic input, leading to elevated network burst activity, and impaired presynaptic vesicle dynamics, causing a lower burst duration in cortical neurons from patient-derived induced pluripotent stem cells harboring C9orf72 repeat expansions (Perkins et al., 2021).Tau:
Tau has both physiological and pathological roles at the presynaptic terminal. In the mutant Tau model of FTD with Parkinsonism-17, Tau binds to the SV protein synaptogyrin-3 to immobilize and prevent SV release. This occurs through Tau binding and crosslinking SVs to the actin cytoskeleton. This crosslinking was shown to reduce synaptic transmission during prolonged neuronal activity (Zhou et al., 2017). Zhou et al. (2017) therefore suggest inhibiting Tau from binding SVs as a possible therapeutic target for FTD linked to MAPT mutations.TDP-43:
TDP-43 in FTD/ALS has already been suggested as a cause of synaptopathy. Its role as an RNA binding protein can affect the expression of mRNAs that encode proteins involved in presynaptic activity. Loss in these synaptic proteins may explain how reduced TDP-43 correlates with impaired synaptic plasticity and synaptic loss (for review see Riku et al., 2021). Decreased release probability and quantal content has also been shown early during pathology of TDP-43Q331K mutation mice, indicating defects in fusion and release of SVs (Chand et al., 2018).FUS:
Like TDP-43, FUS is also an RNA binding protein, and its target RNAs include those associated with synapse organization and plasticity. Interestingly, super resolution microscopy shows that synaptic FUS predominantly localizes near the reserve pool of SVs (Sahadevan et al., 2021). Studies in ALS causative mutant FUS have shown severely impaired neurotransmitter release, quantal size, and altered presynaptic active zone structure in Drosophila models, resulting in severely decreased evoked synaptic transmission, as well as decreased spontaneous miniature excitatory potentials (Shahidullah et al., 2013).Perspective:
CHMP2B FTD is a novel synaptopathy, characterized by defective SV trafficking and retention of select SVassociated proteins, despite significant synaptic and neuronal loss. Through other genes that are involved in ALS/FTD, namely C9orf72, Tau, TDP-43, and FUS, it appears that disruption to synaptic cytoskeleton and exocytosis are common factors that may cause synaptic impairment and reduced synaptic transmission (Figure 1)
.Following exocytosis, multiple pathways are available for synaptic vesicle retrieval and reformation; kiss-and-run, clathrin-mediated endocytosis, activity-dependent bulk endocytosis, and direct ultrafast endocytosis. Interestingly, these diverse modes of retrieval alter the molecular identity and subsequent physiological role of the resultant vesicle (Chanaday et al., 2019), indicating that the different retrieval pathways may impact disease differentially, and thus should be investigated in further detail.
As shown inFigure 1
, there are multiple points of the SV cycle where ALS/FTD associated proteins have been shown to impact on SV exo- and endocytosis. However, it is not yet known what initiates this cascade of SV cycling dysfunction. Identification of a common pathomechanism leading to this SV dysfunction and synaptopathy in FTD/ALS offers the possibility of a common therapeutic target relevant to multiple forms of genetic disease. Evidence for presynaptic synaptopathy as described above comes from multiple model systems using different investigative approaches. It would be extremely interesting to conduct experiments investigating the SV cycle in directly comparable systems, for example investigating cortical and motor neurons derived from CRISPR mediated knock-in of disease relevant mutations on an isogenic induced pluripotent stem cell background for a direct comparison. This would provide direct evidence for a convergent pathomechanism of SV cycle dysfunction preceding neuron death in FTD/ALSThough we have focused here on pathological presynaptic mechanisms, mutations in many of these genes have been associated with both pre- and post-synaptic alterations, which may occur simultaneously or sequentially due to lack of functional signaling. Additionally, while we have focused on the evidence for cell-autonomous mechanisms of SV cycle defects in FTD/ALS, it should be noted that non-cell-autonomous mechanisms of synaptic dysfunction are also likely contributors to synaptopathy, for example, C9orf72 is required for microglial function in mice (O’Rourke et al., 2016).
FTD/ALS synaptopathy is an early event in neurodegeneration that contributes significantly to disease etiology prior to neuron loss. This strongly affirms that targeting the molecular pathways involved in SV trafficking is a promising route to treat FTD/ALS, underlining the importance of further understanding this process and the mechanisms through which synaptopathy leads to neuronal death in FTD/ALS.
MR was funded by MRC LMB. EC acknowledges funding from Alzheimer’s Research UK (PPG2018B-017) and the UK Dementia Research Institute which receives its funding from DRI Ltd., funded by the UK Medical Research Council, Alzheimer’s Society, and Alzheimer’s Research UK.
No conflicts of interest exist between DRI Ltd. and publication of this paper.
Miranda Robbins, Emma L. Clayton
MRC Laboratory of Molecular Biology; Department of Zoology, University of Cambridge, Cambridge, UK (Robbins M)
UK Dementia Research Institute at King’s College London; Department of Basic and Clinical
Neuroscience, Institute of Psychiatry, Psychology and Neuroscience, King’s College London, Maurice Wohl Clinical Neuroscience Institute, London, UK (Clayton EL)
Correspondence to:
Emma L. Clayton, PhD, emma.clayton@kcl.ac.uk.https://orcid.org/0000-0003-0937-2874(Emma L. Clayton)
Date of submission:
January 5, 2022Date of decision:
February 8, 2022Date of acceptance:
February 25, 2022Date of web publication:
July 1, 2022https://doi.org/10.4103/1673-5374.343905
How to cite this article:
Robbins M, Clayton EL (2023) Synaptopathy in CHMP2B frontotemporal dementia highlights the synaptic vesicle cycle as a therapeutic target. Neural Regen Res 18(2):315-316.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:
Devesh Pant, Emory University School of Medicine, USA.
Additional file:
Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- c-Abl kinase at the crossroads of healthy synaptic remodeling and synaptic dysfunction in neurodegenerative diseases
- The mechanism and relevant mediators associated with neuronal apoptosis and potential therapeutic targets in subarachnoid hemorrhage
- Microglia depletion as a therapeutic strategy: friend or foe in multiple sclerosis models?
- Brain and spinal cord trauma: what we know about the therapeutic potential of insulin growth factor 1 gene therapy
- Functions and mechanisms of cytosolic phospholipase A2 in central nervous system trauma
- Cre-recombinase systems for induction of neuronspecific knockout models: a guide for biomedical researchers