
c-Abl kinase at the crossroads of healthy synaptic remodeling and synaptic dysfunction in neurodegenerative diseases
2022-06-29 08:14DanielaGutirrezAmricaChandCristiMarJosezSilvanaZanlungoAlejandralvarez
中国神经再生研究(英文版) 2023年2期
Daniela A. Gutiérrez, América Chandía-Cristi, María José Yáñez, Silvana Zanlungo, Alejandra R. Álvarez,
Abstract Our ability to learn and remember depends on the active formation, remodeling, and elimination of synapses. Thus, the development and growth of synapses as well as their weakening and elimination are essential for neuronal rewiring. The structural reorganization of synaptic complexes, changes in actin cytoskeleton and organelle dynamics, as well as modulation of gene expression, determine synaptic plasticity. It has been proposed that dysregulation of these key synaptic homeostatic processes underlies the synaptic dysfunction observed in many neurodegenerative diseases. Much is known about downstream signaling of activated N-methyl-D-aspartate and α-amino-3-hydroxy-5-methyl-4-isoazolepropionate receptors; however, other signaling pathways can also contribute to synaptic plasticity and long-lasting changes in learning and memory. The non-receptor tyrosine kinase c-Abl (ABL1) is a key signal transducer of intra and extracellular signals, and it shuttles between the cytoplasm and the nucleus. This review focuses on c-Abl and its synaptic and neuronal functions. Here, we discuss the evidence showing that the activation of c-Abl can be detrimental to neurons, promoting the development of neurodegenerative diseases. Nevertheless, c-Abl activity seems to be in a pivotal balance between healthy synaptic plasticity, regulating dendritic spines remodeling and gene expression after cognitive training, and synaptic dysfunction and loss in neurodegenerative diseases. Thus, c-Abl genetic ablation not only improves learning and memory and modulates the brain genetic program of trained mice, but its absence provides dendritic spines resiliency against damage. Therefore, the present review has been designed to elucidate the common links between c-Abl regulation of structural changes that involve the actin cytoskeleton and organelles dynamics, and the transcriptional program activated during synaptic plasticity. By summarizing the recent discoveries on c-Abl functions, we aim to provide an overview of how its inhibition could be a potentially fruitful treatment to improve degenerative outcomes and delay memory loss.
Key Words: actin cytoskeleton; activity-dependent plasticity; Alzheimer’s disease; c-Abl; dendritic spines; learning; synapse; synaptic plasticity; transcription; tyrosine kinase

Introduction 237 Search Strategy 237 Activity-Dependent Signaling Entails Synaptic Plasticity 237 c-Abl Structure and Functions 238 c-Abl Overactivation Contributes to Synaptic Dysfunction and Elimination in Neurodegenerative Diseases 240 Summary 242
Introduction
In the central nervous system, neurons communicate through synapses, highly dynamic structures that, upon stimulation, allow the transmission of information. The synaptic compartment includes actin enriched structures known as dendritic spines, characterized by a pre-synaptic region that contains neurotransmitter vesicles and an electron-dense zone called the post-synaptic density (PSD) that anchors neurotransmitter receptors (Kim et al., 2004). Most hippocampal synapses are glutamatergic in which chemical neurotransmitters are released from vesicles at the pre-synaptic terminal along axons, and received by corresponding N-methyl-D-aspartate receptors (NMDAR) and α-amino-3-hydroxy-5-methyl-4-isoazolepropionate receptors (AMPAR) at the dendritic post-synaptic compartment. The heads of glutamatergic dendritic spines contain mostly NMDAR and AMPAR, PSD-95 and SAP102 scaffold proteins, cellular organelles, coated vesicles, and cytoskeleton filaments, and polyribosomes for protein synthesis (Sabatini and Svoboda, 2000).
Long-term potentiation (LTP) and long-term depression (LTD) are forms of Hebbian plasticity, the bases of learning and memory that underlie the remodeling of synaptic complexes, changes in the actin cytoskeleton, organelle dynamics, and modulation of gene expression; all processes that can be altered by homeostatic mechanisms (Galanis and Vlachos, 2020). After neuronal stimulation, many kinases and phosphatases orchestrate synaptic plasticity (Woolfrey and Dell’Acqua, 2015). However, there is still much to be learned about the role of phosphorylation modulating mechanisms that control synaptic plasticity. It has been proposed that dysregulation of these key synaptic homeostasis processes underlies the synapse dysfunction characteristic of many neurodevelopmental disorders and neurodegenerative diseases. In this review, we summarized recent studies showing how c-Abl tyrosine kinase could control homeostatic plasticity through actin cytoskeleton regulation of dendritic spine morphology, and transcription, to alter learning and memory. We will focus on how these different mechanisms could affect neurodegenerative diseases, specifically Alzheimer’s, Parkinson’s, and lysosomal storage diseases.
Search Strategy
This review analyzes studies found on the PubMed database using the following keywords: c-Abl tyrosine kinase, synaptic plasticity, activitydependent transcription, actin cytoskeleton, dendritic spines, and neurodegenerative diseases. All studies cited were published between 1992 and 2022, and represent the most relevant articles in the field.
Activity-Dependent Signaling Entails Synaptic Plasticity
Synapses continuously form and remodel as part of activity-dependent changes in neuronal connections: a process is known as Hebbian synaptic plasticity, which can be altered by homeostatic compensatory mechanisms to restore network functions (Styr and Slutsky, 2018; Galanis and Vlachos, 2020). Hebbian plasticity involves increasing or decreasing the strength of glutamatergic synaptic transmission, mainly known as LTP and LTD respectively, which allows the functional transmission of information, and it is thought to underlie learning and memory (Bin Ibrahim et al., 2022). The early phase of LTP (1–2 hours) depends on AMPAR localization at the PSD; activation of NMDAR subunits GluN2A/B, and the membrane-associated guanylate kinases comprising PSD-95, PSD-93, and SAP102, which are highly expressed at excitatory synapses where the receptors are anchored (Chen et al., 2021). During LTP, local protein synthesis is ubiquitous in the pre-and post-synaptic compartments and starts rapidly using mRNAs already located at the synapse (Hafner et al., 2019). Late-LTP (weeks to months) relies on Ca-calmodulin proteins (i.e., CaMKII) that couple synaptic activity with the nucleus, promoting the transcription of activity-dependent genes for thede novo
protein synthesis required for the generation of memory (Yap and Greenberg, 2018).LTD involves endocytosis and trafficking of receptors that interchange location, structural reorganization of PSD complexes, and degradation of organelles and synaptic components (Rajgor et al., 2021). The elimination of LTD causes deficits in learning and memory through the dysregulation of AMPAR trafficking (Hanley, 2014). The AAAATPase Thorase mediates surface internalization of AMPAR by disassembling the AMPAR-GRIP1 complex, therefore affecting glutamatergic transmission. Loss of Thorase enhances LTP and impairs LTD by impeding AMPAR endocytosis (Zhang et al., 2011;Figure 1
).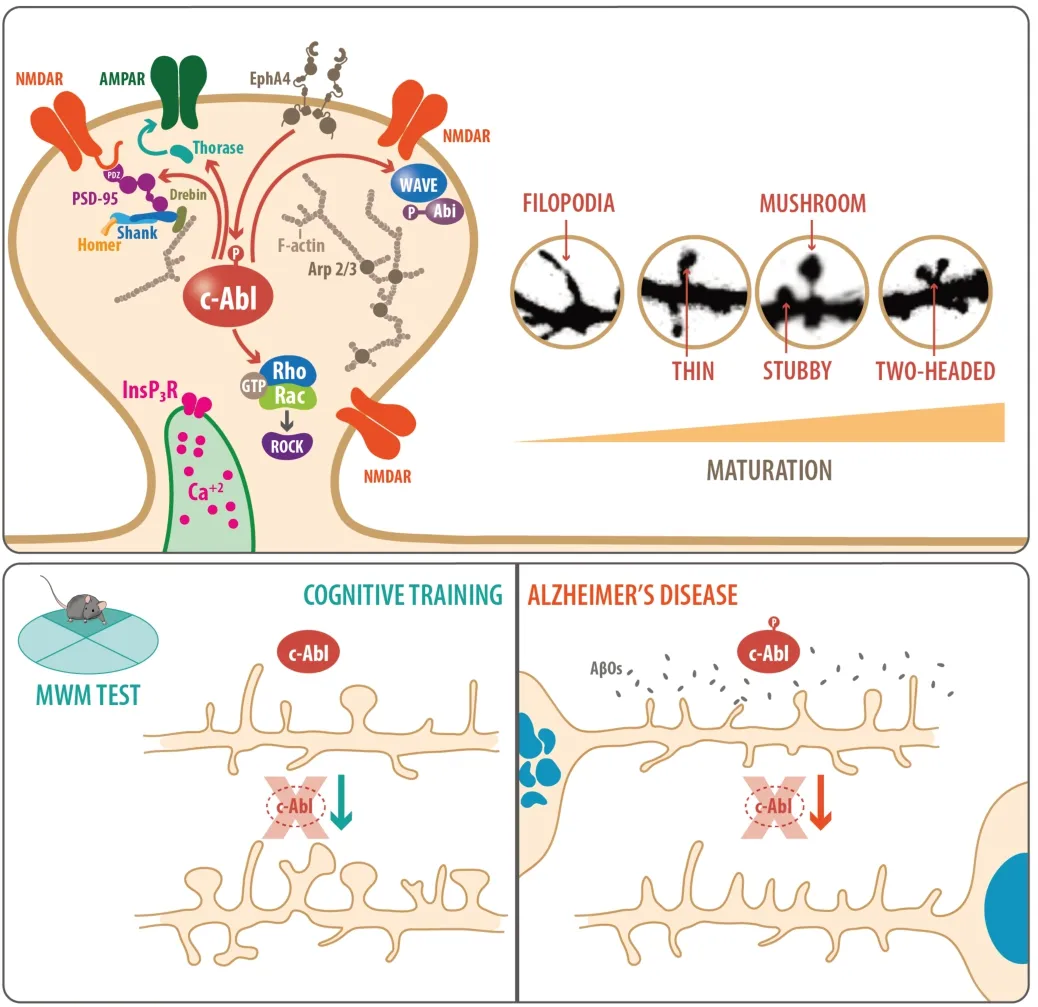
Figure 1|Influence of c-Abl on dendritic spines. Upper panel: Dendritic spines are synaptic compartments with an electron dense zone, the post-synaptic density (PSD), mostly containing PSD-95 and Shank. N-methyl-Daspartate receptors (NMDAR) and α-amino-3-hydroxy-5-methyl-4-isoazolepropionate receptors (AMPAR) are attached to the PSD through PDZ domains. Ephrin A4 receptors respond to Aβ-oligomers (AβOs) binding, activating c-Abl to promote neurodegeneration. EphA4, PSD-95 (Y533), F-actin associated proteins Rho/Rac, Rho-associated protein kinase (ROCK), WASP-family verprolin-homologous protein (WAVE) (Y150) and Abi are regulated by c-Abl. Shape changes during maturation are illustrated by confocal microscopy sections of dendritic spines: filopodia (most immature), thin, stubby, mushroom (most mature), branched, or two-headed spines. Bottom panels: c-Abl alters spine density and induces a shift in the morphology of dendritic spines. c-Abl knockout (c-Abl-KO) mice subjected to cognitive training (MWM: Morris water maze) show increased spine density and display hippocampal neurons that are enriched in mature forms such as mushroom spines (left). AβOs-induced synaptotoxicity decreases spine density independent of c-Abl, and promotes enlargement of spines increasing filopodia, especially in c-Abl-KO neurons. AβOs normally induce apoptosis, but the absence of c-Abl prevents it (right).
Multiple kinases and phosphatases orchestrate synaptic plasticity through the rapid phosphorylation/dephosphorylation of targets that further amplifies signals to the nucleus to promotede novo
translation of proteins (Woolfrey and Dell’Acqua, 2015). For example, after neuronal stimulation, CaMKIIα phosphorylates NMDAR GluN2B and GluN2A subunits promoting endosomal recycling of the receptor (Yong et al., 2021). CaMKIIα also phosphorylates the GluA1 subunit of AMPAR, increasing single-channel conductance and synaptic incorporation promoting LTP (Kristensen et al., 2011). Downstream NMDAR, the activation of CaMKII and protein kinase C increases the levels of the AMPAR in the synapse enhancing synaptic strength (Kristensen et al., 2011). Altogether, these functions made CaMKII a promoter of Hebbian plasticity.Fyn, a member of the Src family of tyrosine kinases, regulates NMDAR localization and trafficking, stabilizing NR2B-containing NMDARs at the synapse and preventing endocytosis (Salter et al., 2004; Trepanier et al., 2012). On the other hand, STEP phosphatase inactivates Fyn and dephosphorylates ERK1/2 promoting NMDAR internalization (Won and Roche, 2021). Therefore, the control of phosphorylation in the synapse becomes essential.
The turnover of dendritic spines during LTP and LTD is a type of structural plasticity. It involves the formation of the synapse, spine morphogenesis, shrinkage, and elimination as part of the activity-dependent restructuration of neuronal connections. Althoughin vivo
imaging studies show that axons and dendrites are quite stable, experience-dependent structural changes do occur (Holtmaat and Caroni, 2016).Dendritic spines arise from the dendritic shaft and are highly dynamic structures. The head of a dendritic spine, which contains the neurotransmitters and scaffold proteins, is also enriched in actin filaments and actin-binding proteins such as Arp2/3, and Rho-Rac/ROCK. These cytoskeletal proteins are responsible for the rapid changes in the shape of the spines during development, response to stimulus, and later maturation (Basu et al., 2018). As dendritic spines undergo maturation their shape changes into five different structures, most commonly known and classified as filopodia, thin, stubby, mushroom, and two-headed spines (Pchitskaya et al., 2020;Figure 1
). The shape and size of dendritic spines determine the strength of the synapse, modifying its morphology by enlargement or shrinkage in response to learning, synaptic activity, environmental enrichment, and damaging stimuli (Fiala et al., 2002; Segal, 2017; Harris, 2020). Filopodia, the nascent spine, is the most immature and longer than 2 µm. It is followed by immature thin spines, and protrusions shorter than 2 µm in length. The intermediate state is the stubby spine, smaller, and neckless. Mature mushroom spines are prominent spines in which the head is wider than the neck (Harris, 2020). It has been suggested that branched or two-headed spines are a consequence of LTP. This type of spine could be induced by an enriched environment and learning after defiant tests in which neuronal activation promotes the maturation of dendritic spines (Figure 1
bottom left panel) (Toni et al., 2001). Thus, it increases the contact area with pre-synaptic axonal terminals and the number of neurotransmitter receptors for the new stimulus, strengthening the synapse.PSD-95 anchors NMDAR to its PDZ domains as a scaffold protein and nascent dendritic spines stabilize as PSD-95 accumulates into the synapse. Overexpression of PSD-95 enhances the maturation of both the pre-and post-synaptic terminals, but also the number and size of dendritic spines (El-Husseini et al., 2000). Spines that do not acquire higher levels of PSD-95 are usually transient, while spines enriched in PSD-95 tend to remain constant in time and mature (Cane et al., 2014).
c-Abl Structure and Functions
c-Abl is a member of the ABL family of tyrosine kinases
The Abelson non-receptor tyrosine kinase (ABL) family includes two members: c-Abl (ABL1), and Arg (Abl-related gene, ABL2) that are ubiquitously expressed in cells. Both share SH2-SH3 (Src homology 2–3) domains, PxxP (Proline-rich) motifs, and F-actin binding domains (BD). Only c-Abl has nuclear localization and nuclear export signals; and a DNA-binding domain that allows nuclear functions related to the regulation of gene expression, DNA damage response, and apoptosis signaling, among others (Colicelli et al., 2010). In contrast, Arg has a microtubule-binding domain (MT-BD) (Figure 2
upper panel). Therefore, both kinases promote changes in cytoskeleton dynamics through direct and indirect interactions.ABL activity is tightly regulated by intramolecular and intermolecular interactions that allow it to adopt active or auto-inhibited conformational states (Figure 2
bottom panel). The auto-inhibited conformation of ABL requires the interaction between the SH3 domain and the linker sequence that connects the SH2 and kinase domains (SH1), leading to the formation of an SH3-SH2 clamp structure that locks and inhibits kinase activity. Additionally, the myristoylated residue in the C-lobe binds to the allosteric hydrophobic pocket of the kinase domain further stabilizing the auto-inhibited conformation. Allosteric inhibitors like GNF-2 bind to the myristoyl pocket preventing activation (Figure 2
bottom panel). Specific phosphorylation of c-Abl Y245 in the linker region between the SH2 and kinase domains prevents reversion to the inactive conformation. Phosphorylation on Y412 in the activation loop of the c-Abl kinase domain orients the catalytic site for substrate phosphorylation (Dorey et al., 2001; Wang et al., 2018). Orthostheric inhibitors such as imatinib and nilotinib compete with ATP preventing phosphorylation (Figure 2
bottom panel). Thus, both Tyr phosphorylations promote c-Abl activity.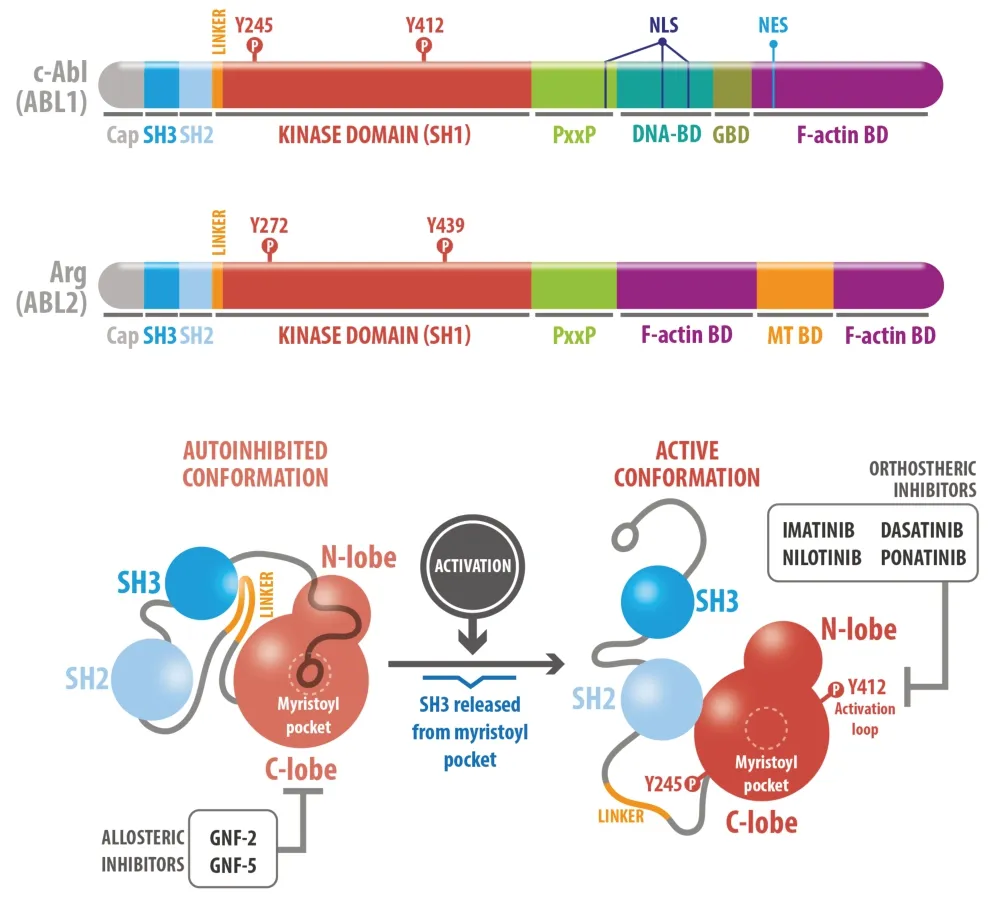
Figure 2|c-Abl structure and enzymatic regulation. Upper panel: Abelson protein tyrosine kinase (ABL) family protein structure: Both, c-Abl and Arg, have an N-terminal kinase domain that starts with a “cap” region, where a myristate can be attached, followed by a highly conserved SH3–SH2–SH1 cassette (homologues to Src-family kinases) and an allosteric pocket. At the C-terminal domain, ABL kinases contain proline-rich sequences (PxxP) that mediate protein-protein interactions due to affinity to SH3-containing proteins and filamentous (F)-actin binding domains (F-actin BD). In particular, c-Abl contains a globular (G)-actin binding domain (GBD) in contrast to Arg, which contains two F-actin and a microtubule-binding domain (MT-BD). Another key difference is that only c-Abl has DNA binding domain (DNA-BD)sequences, nuclear export sequences (NES), and nuclear localization signals (NLS). Bottom panel: SH3 domain binds into the allosteric site of the C-lobe, the myristoyl pocket, and generates an autoinhibited conformation, enhanced by allosteric inhibitors GNF-2 and GNF-5. Release of the SH2-SH3 domain by phosphorylation of Y245 and Y412 of the kinase domain promotes the active conformation. Orthostheric inhibitors imatinib, ponatinib, nilotinib, and dasatinib bind to the activation loop.
ABL kinases are key transducers of signals from DNA damage and oxidative stress, as well as from growth factors and axon-guidance receptors, among other signals. Their substrates are functionally diverse, including adaptors, other kinases, cytoskeletal proteins, transcription factors, chromatin modifiers, and others. Despite ABL kinases’ ubiquitous expression and multiple functions in different cell types, their nervous system-associated roles and participation in neurological diseases, have promoted them as emerging therapeutic targets. c-Abl and Arg are expressed throughout the adult mouse brain. Interestingly, ABL kinases are highly expressed in neurons, especially in pre- and post-synaptic terminals, and their activity regulates the reorganization of the cytoskeleton associated with dendrogenesis (Jones et al 2004; Shaw et al., 2021). Most studies on ABL kinases in the neuronal synapse evaluate the role of Arg using Argdeficient mice (Lin et al., 2013). For example, integrin-regulated Arg kinase coordinates the maturation of pre- and postsynaptic compartments by allowing low-release probability GluN2B synapses to replace high-release probability GluN2B synapses during development. This turnover is critical for synaptic plasticity (Xiao et al., 2016). Also, Arg dampens activity-dependent disruption of cortactin localization to stabilize spines and attenuates Rho activity to stabilize dendrite arbors (Lin et al., 2013). There are a few studies about the role of c-Abl in synaptic structure since c-Abl has been mostly associated with neurodegenerative diseases because of its participation in altered regulation of cytoskeleton dynamics, gene expression, and apoptosis (Vargas et al., 2018). As we describe here, compelling evidence places c-Abl as one of the main regulators of the most important molecular pathways that regulate the synapse and neuronal homeostasis during synaptic plasticity.
c-Abl signaling and neuronal cytoskeleton dynamics
One of the main biological functions of ABL tyrosine kinases is to control actin cytoskeleton remodeling. F-actin is required for the proper development and organization of pre- and post-synaptic components and the formation of cellular structures such as lamellipodial protrusions, filopodia, and dorsal membrane ruffles. c-Abl regulates these processes, which are fundamental for neurite formation and branching, synaptogenesis, and synaptic plasticity (Woodring et al., 2003; Zhang et al., 2018; Gonzalez-Martín et al., 2021).
Multiple experiments using primary cultures of hippocampal neurons have shown that c-Abl modulates neuronal morphogenesis, dendrite outgrowth, and branching. It is also known that c-Abl induces neurite extension by modulating actin cytoskeleton dynamics, either directly through its F-actin BD or indirectly through phosphorylation of actin-binding proteins such as the small GTPase RhoA (Woodring et al., 2003; Jones et al., 2004), and Cdk5 (Zukerberg et al., 2000; Cancino et al., 2011). c-Abl also regulates actin polymerization by phosphorylating Abl-interactor 1 (Abi) and WAVE2 on Tyr150, components of the WAVE regulatory complex necessary for the activation of Arp2/3 (Bradley and Koleske, 2009; Mendoza, 2013;Figure 1
). On the other hand, Arg regulates both the actin and microtubule cytoskeletons downstream from the activation of axon guidance receptors to modulate the guidance of axonal growth cones (Clarke et al., 2020). Recently, studies have revealed functional interactions between the Arg C-terminus and microtubules, allowing direct regulation of MT dynamics (Hu et al., 2019).Many molecular pathways affect dendritic spine stability. However, here we summarized those related to c-Abl (Table 1
). The main actin-binding proteins controlling dendritic spine dynamics are Arp2/3, which promote actin polymerization; the GTPases RhoA/Rac1; and kinase ROCKII (Table 1
). Activation of RhoA leads to dendritic spine loss, while its inhibition promotes spinogenesis of long and motile spines (Tashiro et al., 2004). It has been reported that c-Abl inactivates RhoA promoting dendrogenesis (Jones et al., 2004). However, there are no studies that focused on the effects of c-Abl on dendritic spines. Rac1 blockade induces long, thin spines, inhibits spine head growth, and decreases spine density (Tashiro et al., 2004). On one hand, c-Abl anchors Rac1 during cell spreading, impeding lamellipodium formation without affecting the activity of Rac1 (Jin and Wang, 2007). On the other hand, Abl-interactor 1 inhibitory interaction with CAMKII inhibits the activation of Rac1 and spine maturation (Park et al., 2012), suggesting that c-Abl could be regulating spine length through Rac1.c-Abl and synaptic complexes
PSD-95 undergoes post-translational modifications, including palmitoylation and serine/threonine phosphorylation that regulates its attachment to NMDAR (Kim et al., 2007). Our group also described that c-Abl co-localizes with PSD-95 and controls its synaptic clustering through Tyr phosphorylation on Y533 (Figure 1
). Inhibition of c-Abl with imatinib and transient genetic ablation of the c-Abl kinase domain (c-Abl-KD) decreases the area of PSD-95 clusters and PSD-95/Synapsin synaptic contacts without variation in the number of dendritic spines (Perez de Arce et al., 2010). However, in mature c-Abl knockout (KO) cultured neurons, the number of PSD-95 clusters was similar to wild-type neurons (Gutierrez et al., 2019), further suggesting that compensatory mechanisms are taking place to ensure synaptic structure. On the other hand, we found that clusters of the pre-synaptic protein Piccolo were significantly enhanced in the absence of c-Abl, while synaptic contacts PSD-95/Piccolo did not vary. Therefore, c-Abl could be differentially influencing pre-synaptic proteins. Since Piccolo is a scaffold protein of the active zone at the presynaptic terminal that maintains the clustering of synaptic vesicles (Gundelfinger et al., 2016), it could be affected by c-Abl regulation of the actin cytoskeleton, which is crucial for vesicle mobilization, trafficking, and neurotransmitter release. However, it has also been shown that c-Abl regulates neurotransmitter release at Schaffer collateral-CA1 synapses (Moresco et al., 2003). Electrophysiological studies showed that c-Abl modulates the efficiency of neurotransmitter release from the pre-synaptic terminal, and c-Abl-KO mice had reduced pulse paired-facilitation (Moresco and Koleske, 2003), further suggesting that c-Abl modulates short-term synaptic plasticity.c-Abl dependent transcription as a regulator of dendritic spine morphology
Interestingly, we recently found that brain-specific c-Abl-KO mice subjected to cognitive training show positive regulation of the actin cytoskeleton and synaptic genes (González-Martín et al., 2021). We observed upregulation of the Actr2 gene and Arp2 protein, which controls dendritic spine dynamics by actin polymerization (Table 1
). The Arp2/3 complex is essential for actinrich filopodial maturation into mushroom spines during spinogenesis, and the recruitment of AMPARs (Spence et al., 2016), suggesting that the enhanced polymerization seen after chemical induction of LTP while c-Abl is inhibited may promote the stabilization of nascent spines. This could explain the increased spine density observed in c-Abl-KO mice after cognitive tests (González-Martín et al., 2021). Therefore, these results suggest that c-Abl participates in synaptic cytoskeletal remodeling during learning.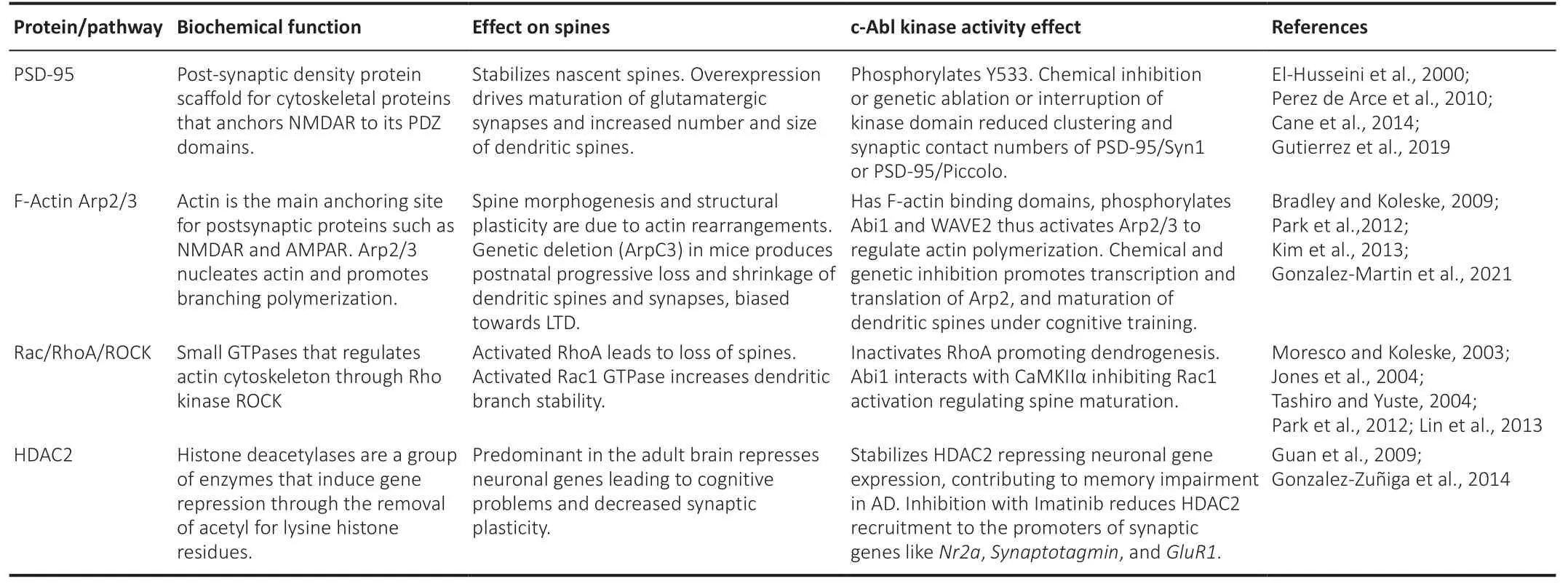
Table 1 | Molecular pathways that affect dendritic spines and c-Abl interactions
After hippocampal-dependent tasks conditioning, c-Abl-KO mice surpass wildtype mice performance and, accordingly, show an enriched spine population with increased head size (mushroom). The bigger dendritic spine head size also increases the contact area with pre-synaptic axonal terminals, and the number of neurotransmitter receptors for a new stimulus, strengthening the synapse. Furthermore, the overall average spine length was increased because of the presence of immature spines (González-Martín et al., 2021;Figure 1
). Thus, c-Abl may contribute to synapse destabilization through the regulation of Arp2/3, deletion of which promotes profound loss of dendritic spines and abnormal filopodial-enriched spine shapes (Kim et al., 2013). Additionally, we found that c-Abl-KO mice showed upregulation of the Atad1 gene and increased Thorase protein expression after 2 days of cognitive training. Thorase is known to participate during LTD induction. Accordingly, Thorase-KO mice show blockade of AMPAR endocytosis; enhanced LTP; and poor performance in open field, Y-maze and T-maze tests, as a result of impaired spatial memory as they are unable to form new memories (Zhang et al., 2011). Therefore, Thorase upregulation in c-Abl-KO mice could contribute to the improved performance and enhanced spatial memory observed (Gonzalez-Martín et al., 2021), as a Hebbian plasticity promoter. It is also important to consider that RIN1 (Ras/Rab interactor 1), through its Rab5 GEF activity (Szíber et al., 2017) in an Abl kinase-dependent pathway, affects the morphology of dendritic protrusions and accelerates filopodia motility destabilizing synaptic connections and enhancing AMPA receptor endocytosis. However, the molecular mechanisms by which c-Abl could be regulating Thorase or even AMPARs recycling remain to be studied.It has been described that c-Abl regulates histone deacetylases (HDACs) activity through phosphorylation. HDACs are a group of enzymes that induce gene repression through the removal of acetyl groups from lysine histone residues. Guan and colleagues showed that the overexpression of HDAC2 highly reduced functional synaptophysin+ synapses and dendritic spine density in the mouse CA1 area of the hippocampus. In contrast, HDAC2 KO mice showed enhanced memory formation in the fear-conditioning memory test and had more spines. HDAC2 represses the expression of synaptic remodeling/plasticity neuronal genes such asBdnf,
Fos
,Camk2a
,Creb
,Nr2a
/b
, among others (Guan et al., 2009). c-Abl kinase activity induces histone deacetylation, in particular, it inhibits proteasomal degradation of HDAC1/2 and stabilizes these complexes in the nucleus of proliferative cells (Aoyama et al., 2015). We showed that c-Abl phosphorylates HDAC2 in Y222 and stabilizes it. c-Abl inhibition reduces HDAC2 levels in neurons and its recruitment to the promoters of synaptic genes likeNr2a
,Synaptotagmin
, andGluR1
(González-Zuñiga et al., 2014;Figure 3
). Therefore, the enhanced transcription of these genes promotes structural plasticity in pre-and post-synaptic terminals.
Figure 3|c-Abl implications in neurodegenerative diseases focusing on Alzheimer’s disease (AD), Parkinson’s disease (PD), and lysosomal storage diseases (LSDs). Summary of cytoplasmic and nuclear functions of c-Abl in neurodegenerative diseases compared with the basal healthy condition. In healthy neurons, different rates of c-Abl kinase activity control the actin cytoskeleton to remodel the synapse. However, it also acts as a brake for transcriptional regulation of cytoskeletal genes like Arp2/3, and synaptic genes like Atad1, which controls AMPARs recycling. c-Abl phosphorylates RNA polymerase II (RNAPII) (Y1) to terminate transcription. In AD, Aβoligomers (AβOs) induce the activation of the Ephrin A4 (EphA4) receptor that activates c-Abl (Y412) to control actin cytoskeleton remodeling and dendritic spine loss. Active c-Abl controls Tau hyperphosphorylation (Y197, Y310, and Y394) through cyclin-dependent kinase 5 (CDK5) activation (Y15), and also phosphorylates p73 (Y99) and Yap1 (Y357) inside the nucleus promoting neuronal apoptosis. Inhibition of c-Abl basally reduces deactylation of histone deacetylaces (HDACs), in particular, it inhibits HDAC2 phosphorylation (Y222) and recruitment to promoters of synaptic genes, favoring transcription. The opposite occurs in AD. In PD, alpha-synuclein (α-syn) aggregates induce aberrant c-Abl activation that phosphorylates synuclein (Y39) and inactivates parkin (Y143), promoting oxidative stress and dysregulation of mitochondrial biogenesis and autophagy. In the LSD Niemman Pick C (NPC) disease, cholesterol accumulation into the lysosomes induces aberrant c-Abl activation that phosphorylates transcription factor EB (TFEB) (Y173), blocks its translocation into the nucleus, and downregulates gene expression. In Gaucher disease (GD), glucosylceramide accumulation into the lysosomes induces aberrant c-Abl activation that further phosphorylates receptorinteracting serine/threonine-protein kinase 3 (RIPK3), which activates mixed lineage kinase domain-like protein (MLKL) and induces necroptosis. Ultimately, c-Abl leads to p73 phosphorylation and neuronal apoptosis. All of these different results point to c-Abl as a target for the development of therapeutic strategies to improve the outcomes of neurodegenerative diseases.
Moreover, c-Abl phosphorylates the CTD-Tyr1 of RNA Polymerase II (RNAPII) during transcription-coupled DNA double-strand breaks (DSBs) in proliferating cells (Burger et al., 2019). Others suggest that RNAPII is poised at activityinduced genes in neurons to rapidly promote transcription through DSBs (Madabhushi et al., 2015), but less is known about RNAPII/c-Abl in neurons.Altogether, these studies indicate that c-Abl affects the synapse, either directly through the actin-cytoskeleton and receptor trafficking, or as a transcriptional regulator of the proteins involved. Therefore, it is a part of the mechanism of homeostatic plasticity. However, due to the various functions that c-Abl fulfills in the cell, understanding how its differential localization and kinase activity rates contribute to dendritic spine remodeling is difficult to understand completely.
c-Abl Overactivation Contributes to Synaptic Dysfunction and Elimination in Neurodegenerative Diseases
Alzheimer’s disease
Alzheimer’s disease (AD) is the main form of dementia in people over 60 years. It affects the brain centers of learning and memory: the entorhinal cortex, and the hippocampus (Viola and Klein, 2015). AD is a neurodegenerative disease characterized by histopathological hallmarks that include the accumulation of senile plaques consisting of aberrant extracellular aggregates of the amyloid-beta (Aβ) peptide and intracellular deposits of hyperphosphorylated tau protein known as neurofibrillary tangles (NFTs) (Hardy and Higgins, 1992; Alonso et al., 2018). c-Abl is activated in AD patients’ brains, andin vitro
andin vivo
AD models (Alvarez et al., 2004; Jing et al., 2009, Olabarria et al., 2019), suggesting a potential role for c-Abl in AD neurodegeneration.Synaptopathogenesis characterizes early AD where Aβ-oligomers (AβOs) bind to the synapse and ultimately promote neuronal death (Fiala et al., 2002; Sciaccaluga et al., 2021). The morphological changes that decrease the strength of the synapse usually found include abnormal filopodial shapes, reduced number, enhanced ectopic spine formation, and shrinkage of dendritic spines (Maiti et al., 2015). Due to Aβ synaptotoxic properties, it has been proposed as a blocker of Hebbian plasticity and promoter of homeostatic synaptic plasticity. However, because of neuronal firing alterations, Aβ is also involved the failure of homeostatic mechanisms in early AD (Styr and Slutsky, 2018; Galanis and Vlachos, 2020).
Our studies showed that the EphA4 receptor binds AβOs; phosphorylates, and causes downstream activation of c-Abl, inducing synaptic loss and LTP blockade. The EphA4 receptor antagonistic peptide KYL or imatinib treatment prevents dendritic spine density loss and improves neuronal survival (Vargas et al., 2014), showing that the Ephrin/c-Abl axis is a key signaling pathway in early AD (Figure 3
) (for further information, see Vargas et al., 2018).In vitro
models for early-AD have shown that c-Abl absence causes the enlargement of the dendritic spine head and increased spine density, as well as maintaining dendritic spine density and promoting the lengthening of the spines while the mushroom population remains (Gutierrez et al., 2019;Figure 1
bottom panel right). Therefore, a transient spine population predominates after AβOs exposure in the absence of c-Abl.Downstream activation of c-Abl with AβOs triggers the phosphorylation and inactivation of the E3A ubiquitin-protein ligase (Ube3A) that regulates the degradation of Arc and Ephexin-5. When these proteins accumulate, surface AMPAR-GluR1 subunit and spine density decrease. Accordingly, c-Abl inhibition overrides this effect (Olabarria et al., 2019). Thus, the resilience against AβOs damage in the absence of c-Abl might be explained by c-Abl regulation of the actin cytoskeleton/Arp2/3, and Ube3A/Arc/Ephexin-5.
c-Abl has nuclear functions that could worsen AD pathology. During long incubation with AβOs, c-Abl kinase activity prompts p73 phosphorylation and the expression of apoptotic genes that trigger neuronal apoptosis (Cancino et al., 2008; Wetzel et al., 2008). Interestingly, the chemical inhibition of c-Abl blocks p73 phosphorylation and decreased HDAC2 recruitment to neuronal promoters (González-Zuñiga et al., 2014;Figure 3
). Therefore, c-Abl promotes HDAC2 transcriptional repression activity and worsens AD neuropathology.Moreover, c-Abl activity has been linked to alterations in tau protein, a classic pathological change in AD. ABL kinases directly phosphorylate tau on Y197, Y310, and Y394; however, only Y197/Y394 is phosphorylated in the NFTs in AD (Tremblay et al., 2010). c-Abl regulates downstream substrates that collectively contribute to tau phosphorylation and NFT formation in AD. For example, Cdk5 activity is elevated in the AD brain, and when deregulated, it contributes to tau hyperphosphorylation and the formation of NFTs (Liu et al., 2016). c-Abl phosphorylates Cdk5 (Y15) and promotes tau phosphorylation in AD transgenic mice (Cancino et al., 2011;Figure 3
). GSK-3β is a constitutively active proline-directed serine/threonine kinase recognized as a key player in AD pathogenesis, since the AD patient’s brain, from pre-tangle to stage VI, displays widespread activated GSK3β (Lauretti et al., 2020). Interestingly, c-Abl phosphorylates GSK-3β on Y216 (Ren et al., 2018), a modification required for its activation. Altogether, these studies suggest that pathological activation of Cdk5 and GSK3β could be mediated by c-Abl in the context of AD.The evidence we discussed above shows that c-Abl has multiple interactions with pathogenic processes related to AD. The abnormal activation of c-Abl modulates cytoskeletal remodeling, synaptic complexes disassembly, and inhibition of plasticity-related gene transcription. We hypothesized that c-Abl acts as a promoter of homeostatic plasticity. The data indicate that c-Abl overactivation due to aberrant processing of homeostatic regulators of neuronal function can cause early synaptic dysfunction and elimination, and ultimately neuronal death.
Finally, the pharmacological manipulation of c-Abl phosphorylation by nilotinib in early AD prevents the degeneration of dopaminergic neurons in the Tg2576 mouse model of AD. Reduction of aberrant c-Abl phosphorylation with nilotinib improves autophagy, reduces Aβ levels, and restores memory deficits (La Barbera et al., 2021).
Nilotinib usage in clinical trials resulted in a reduction of amyloid burden in the frontal lobe. Participants with mild to moderate AD that were orally treated with nilotinib had also lower Aβlevels in the cerebrospinal fluid (Turner et al., 2020). This study further supports the positive effects of c-Abl inhibitors and their tolerability for the treatment of AD.
Lysosomal storage diseases
Lipids are essential components of plasma and organelle membranes that serve as a reservoir for intracellular molecules participating in physiological and pathological functions in the brain. For example, cholesterol is a key component of dendritic spines and participates in synaptic plasticity and the induction of LTP (Koudinov and Koudinova, 2001). Although the molecular mechanisms by which cholesterol modulates synaptic plasticity are not clear, changes in the PI3K/Akt pathway, inactivation of GSK3β, and reduced AMPAR internalization have been described (Martin et al., 2014).
Alterations in lipid metabolism have been associated with neurodegenerative disorders. For example, animal models of lysosomal storage diseases (LSDs), including mucopolysaccharidosis type IIIC, Sanfilippo disease type C, and Tay-Sachs’ disease, show synaptic dysfunction, abnormal spine morphology (immature and reduced number of dendritic spines), and electrophysiological deficits (Dwyer et al., 2017; Pará et al., 2020). Furthermore, indirect evidence connects alterations in lipid metabolism with changes in synaptic plasticity in Niemann-Pick disease type A, Niemann-Pick disease type C (NPC), and Gaucher disease (GD), in which lysosomal function is impaired (Rebiai et al., 2021). NPC disease results from defects in cholesterol transport from the lysosome, and as a result, cholesterol is accumulated in this compartment. NPC neurons develop significant alterations in the pre-synaptic structure and function with important neuronal damage, loss, and LTP impairment (D’Arcangelo et al., 2016). Recent discussions focus on how a given lysosomal deficiency triggers synaptic dysfunction and how lysosomal storage defects result in changes in the structure and function of the synapse (see Rebiai et al., 2021 for further information).
Padamsey et al. (2017) demonstrated that dysfunction in Calysosomal signaling and aberrant lysosomal fusion with the plasma membrane might contribute to the loss of dendritic spines and neurons in NPC. Moreover, Tiscione et al. (2019) showed that NPC1 inhibition or disease causing mutations reduced endoplasmic reticulum Calevels by presenilin-1 dependant-Caleak. The dysregulation in calcium homeostasis in NPC1 animals destabilizes neuronal dendritic spines (Tiscione et al., 2019). Interestingly, inhibition of the sterol-response element-binding protein pathway restores Cahomeostasis, reduces the NPC1-characteristic accumulation of cholesterol, and rescues spine density (Tiscione et al., 2019). Neurological manifestations in LSDs share multiple pathophysiological mechanisms with other neurodegenerative diseases such as AD, including the proteins that are implicated: tau, presenilins, and Aβ (Almeida et al., 2020).In vitro
andin vivo
models of NPC had altered expression of APP, whose amyloidogenic processing can be reduced by inhibiting c-Abl (Yáñez et al., 2016). Recent studies show that c-Abl signaling is key to promoting neuronal death in different neurodegenerative diseases, including LSDs.We previously reported the activation of c-Abl and its pro-apoptotic target, p73, in the cerebellum of NPC mice. However, the upstream signals underlying the engagement of this pathway remain unknown (Alvarez et al., 2008). Using differentin vitro
andin vivo
NPC models, we investigated the possible role of oxidative stress in the activation of c-Abl/p73. Our results indicate a close temporal correlation between the onset of oxidative stress and the activation of c-Abl/p73 in NPC models (Klein et al., 2011). Moreover, c-Abl activity increases HDAC2 levels inducing neuronal gene repression of key synaptic genes in NPC models (Contreras et al., 2016). Furthermore, active c-Abl induces the phosphorylation of Tyr and the retention of the transcription factor TFEB in the cytosol, which induces the expression of genes involved in lysosomal biogenesis and exocytosis, and autophagy. Accordingly, c-Abl inhibition promotes TFEB nuclear localization and the expression of its target genes, inducing lysosomal biogenesis, exocytosis, autophagy, and promoting cholesterol clearance in NPC models (Ren et al., 2018; Contreras et al., 2020;Figure 3
). On the other hand, we found that aberrant phosphorylation of c-Abl is increased in several NPC and GD models (Yáñez et al., 2016, 2021). GD is caused by mutations in the GBA1 gene that encodes for the lysosomal enzyme β-Glucocerebrosidase leading to glucosylceramide accumulation in lysosomes. We found that c-Abl mediates the activation and phosphorylation of the RIPK3 serine/threonine kinase (Yáñez et al., 2021), which is involved in the RIP1/RIP3/MLKL necroptotic pathway (Figure 3
). Despite the information presented here on c-Abl substrates involved in LSDs outcomes, there is no published information on the role of c-Abl on the synapse in LSDs. Luckily, some of our data were published during the reviewer’s revision of this work. We showed that Niemann-Pick disease type A mice subjected to the memory flexibility learning test for spatial memory recall improved cognitive performance when fed a diet supplemented with c-Abl inhibitors (Marin et al., 2022). Furthermore, these treated Niemann-Pick disease type A mice also showed delayed locomotor deterioration, less loss of cerebellar Purkinje neurons, and decreased neuronal disorganization in the CA1-CA3 hippocampal circuit (Marin et al., 2022). Our work suggests that increased autophagosome-lysosome fusion restored autophagy flux after inhibition of c-Abl, decreasing neuronal death.All these results provide new evidence for understanding how c-Abl contributes to LSDs pathogenic mechanisms and position c-Abl as a therapeutic target for the treatment of patients with diseases in which the lysosomes are compromised.
As Rebiai et al. (2021) state, an interplay of factors such as the accumulation of undegraded substrates within the synaptic endo-lysosomal pathway, altered lysosomal structure and function, autophagy impairment, and c-Abl activation and function, would contribute to synaptic alterations in LSDs. However, much is still to be investigated on how c-Abl participates in synaptic dysfunction in LSDs.
Parkinson’s disease
Parkinson’s disease (PD) is characterized by misfolded α-synuclein aggregates that degenerate dopaminergic neurons in the substancia nigra pars compacta. c-Abl is an important pathogenic mediator of the disease, where activated c-Abl emerges as a common link to various PD-related inducers of oxidative stress relevant to sporadic and familial forms of PD and other α-synucleinopathies (Brahmachari et al., 2016). c-Abl directly phosphorylates α-synuclein in Tyr39 inducing its aggregation. c-Abl inhibition with nilotinib promotes α-synuclein clearance via the autophagy and proteasome pathways (Mahul-Mellier et al., 2014). α-Synuclein inclusions cause a major reduction in neuronal connectivity, synchronicity, and excitatory tone (Volpicelli-Daley et al., 2011). These aggregates can disrupt LTP in striatal spiny projection neurons, and induce a reduction in surface levels of GluN2A-NMDAR, leading to decreased NMDAR currents. Therefore, aberrant c-Abl activation that promotes α-synuclein aggregation, could play a role in targeting NMDA/AMPA receptors to the cell surface, directly affecting synaptic transmission and plasticity in PD.
In the MPTP mouse model of PD, nilotinib ameliorates motor deficits by normalizing altered activity in postsynaptic signaling pathways in the striatum. Specifically, prevents the increase of phosphorylated Cdk5-Tyr15 and DARPP-32-Thr75 mediated by c-Abl (Tanabe et al., 2014), supporting the detrimental role of c-Abl in synaptic transmission. Also, the c-Abl-GSK3β pathway mediates autophagy by inhibiting TFEB nuclear localization with the subsequent neuronal loss in primary midbrain neurons treated with MPP(Ren et al., 2018).
Overexpression of the constitutively active form of c-Abl (CA) accelerates neurodegeneration through the accumulation of ubiquitinated proteins, which exacerbates behavioral deficits and reduces the lifespan of hA53T α-synuclein transgenic mice (Brahmachari et al., 2016). Moreover, c-Abl-CA overexpression is sufficient for the degeneration of dopaminergic neurons in the substancia nigra of wild-type mice. In contrast, c-Abl-KO increases the survival of hA53T mice and delays behavioral, neurodegenerative, and pathological features (Brahmachari et al., 2016).
c-Abl also phosphorylates parkin in Tyr143, which leads to the disruption of its E3-ligase enzymatic activity and the impairment of parkin-dependent pathways for protein clearance of phosphorylated/internalized α-synuclein aggregates or plaques (Ko et al., 2010;Figure 3
). Hence, c-Abl mediated inactivation of parkin leads to downregulation of ubiquitination of different parkin substrates that contribute to exacerbating neurodegeneration of dopaminergic neurons.Accordingly, treatment with c-Abl inhibitors or genetic deletion of c-Abl suppresses the effects on parkin and α-synuclein, promoting protein clearance and protecting against neurodegeneration.
Given that activated c-Abl is markedly increased in brain samples from PD patients and c-Abl inhibitors treatment shows a consistent neuroprotective phenotype in model studies, it is possible to consider c-Abl inhibition as a therapy for PD. However, recent studies using nilotinib have been unsuccessful for PD patients, apparently due to poor drug availability in the brain (Pagan et al., 2020).
Summary
In this review, we mentioned different functions of the non-receptor tyrosine kinase c-Abl (ABL1) to elucidate how its activation in the brain affects learning and memory. We analyzed ABL kinases structure to highlight the differences between Arg and c-Abl on its actin and DNA-binding domains, and nuclear localization signals that allow c-Abl to respond to a wide variety of stimuli. We embarked on our task into Hebbian and homeostatic synaptic plasticity by reviewing different molecular pathways that participate in altering the morphology of dendritic spines. These structures attract the special attention of scientists for being the center of learning and memory, as first described by Santiago Ramón y Cajal. Alterations of dendritic spines’ shape, size, and density, are usually found in neurodegenerative diseases. However, not every change in the shape and number of the dendritic spines is detrimental, as they also occur during synaptic plasticity. In this context, in absence of c-Abl, the dendritic spine’s head widens, and their number increases in response to activity-dependent plasticity or LTP after conditioned learning, as we hypothesized, acting as a promoter of Hebbian synaptic plasticity. However, c-Abl overactivation contributes to worsening the outcome of neurodegenerative diseases, acting as a homeostatic mechanism of synaptic plasticity.
Finally, we analyzed c-Abl involvement in AD, PD, and LSD to emphasize that pharmacological inhibition of c-Abl with small and central nervous system permeable inhibitors could be a valuable strategy for treating the cognitive decline and memory impairments characteristic of these diseases. Therefore, we perceive a therapeutic window of opportunity for the treatment of neurodegeneration that will encourage new studies to further comprehend the role of c-Abl in the regulation of synaptic plasticity.
Acknowledgments:
We thank Felipe Serrano from Illustrative Science, Santiago, Chile for his art work to make the figures.
Author contributions:
DAG wrote the section of synaptic plasticity, c-Abl on dendritic spines in learning, Parkinson, and Alzheimer’s disease. ACC wrote the section of c-Abl structure, cytoskeleton-associated proteins, and Alzheimer’s disease. MJY wrote the section of lysosomal storage diseases and c-Abl. ARA and SZ helped to write and edit all sections. All authors approved the manuscript.
Conflicts of interest:
The authors declare no conflicts of interest.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:
Han Seok Ko, Johns Hopkins University School of Medicine, USA.

- 中国神经再生研究(英文版)的其它文章
- The mechanism and relevant mediators associated with neuronal apoptosis and potential therapeutic targets in subarachnoid hemorrhage
- Microglia depletion as a therapeutic strategy: friend or foe in multiple sclerosis models?
- Brain and spinal cord trauma: what we know about the therapeutic potential of insulin growth factor 1 gene therapy
- Functions and mechanisms of cytosolic phospholipase A2 in central nervous system trauma
- Cre-recombinase systems for induction of neuronspecific knockout models: a guide for biomedical researchers
- Prenatal programing of motivated behaviors: can innate immunity prime behavior?