
SNRPA1基因在胃癌中的表达及对胃癌细胞生长的作用※
2022-06-22 02:29:16陈思嫒孙静薇苏占海
中国高原医学与生物学杂志 2022年3期
陈思嫒,孙静薇,刘 雅,安 娟,苏占海
(青海大学医学院基础医学部,中藏药抗肿瘤基础与应用研究实验室,青海 西宁 810016 )
最新的研究认为[1],剪接体与癌症进展相关。SNRPA1(small nuclear ribonucleoprotein polypeptide A 1)作为主要剪接体成分—U2核小核糖核蛋白(U2 small nuclear ribonucleo protein,U2 snRNP)复合体的重要剪接因子,在剪接体的组装、获取和维持方面起重要作用[1]。SNRPA1在胃癌发生发展中的作用尚不明确。在前期实验中,我们结合TCGA分析胃癌差异表达谱发现SNRPA1在胃癌中高表达,进一步通过高通量差异基因增殖筛选技术(High Content Screening,HCS)发现干扰SNRPA1基因可以抑制胃癌细胞的生长。因此我们提出剪接体相关蛋白SNRPA1可能会影响胃癌细胞表达的假设,拟通过生物信息学及肿瘤细胞生物学实验分析、探讨SNRPA1在胃癌发生发展中的作用。
1 材料与方法
1.1 主要材料、仪器
胃癌细胞系NCI-N87、MKN-74、MKN-45、HGC-27、KATO III、AGS(由北京肿瘤医院肿瘤分子学实验室惠赠),胎牛血清(ExCell Bi0),1%青霉素-链霉素混合液(Solarbio),DMEM基础高糖培养基(Procell),胰蛋白酶-EDTA消化液(Solarbio),嘌呤霉素(Solarbio),总RNA提取试剂盒(TIANGEN),FastKing gDNA Dispelling RT SuperMix(TIANGEN),SuperReal PreMix Color(TIANGEN),CCK-8试剂溶液(Elabscience),PBS(Solarbio),4%多聚甲醛(Leigen),吉姆萨染液(Solarbio)。
NanoDrop 2000紫外分光光度计(Thermo),实时荧光定量PCR仪(Thermo)。
1.2 方法
1.2.1SNRPA1基因表达水平差异性的分析方法
利用GEPIA2 (http://gepia2.cancer-pku.cn)在线分析工具对SNRPA1基因表达水平进行差异性分析,筛选数据库胃癌组织408例、癌旁组织36例,统计量参数“log2FC cut off”设置为1、“q-value cut off”设置为0.01、“Log Scale”设置为No。使用ANOVA单因素方差统计法分析相关数据,当log2FC>1时,说明该基因在胃癌中表达较高;<1时,说明该基因在胃癌中表达较低。以log2|Fold Change|>2及校正P<0.05作为判断表达基因的差异标准。
1.2.2SNRPA1在不同癌症中的转录水平的分析方法
利用Oncomine(https://www.oncomine.org)在线数据集分析关键基因SNRPA1在不同癌症中的转录水平,将癌组织中的基因mRNA表达水平与正常水平比较,采用t检验,生成P值。P值和折叠变化值的界限分别定义为0.01和2。
1.2.3 探索SNRPA1在胃癌中的分子作用机制的方法
为了进一步探索特征基因SNRPA1在胃癌中的分子作用机制,我们进行了蛋白互作网络分析,利用STRING(https://string-db.org)数据库(数据来自ELIXIR数据库)将与特征基因SNRPA1相关的105个基因建立互作网络,下载PPI网络数据并导入CytoScape软件(mac版3.8.2)中,进行PPI网络(protein protein interaction network,PPI network)分析。
1.2.4 胃癌细胞培养方法
将冻存的胃癌细胞系NCI-N87、MKN-74、MKN-45、HGC-27、KATO III、AGS置于37 ℃水浴锅中快速融化,转移至用10%胎牛血清、1%青霉素-链霉素混合液、DMEM基础高糖培养基配制的完全培养基中入恒温细胞培养箱(37℃,5%CO2)培养。24 h后更换新鲜完全培养基。当细胞密度大于80%且细胞生长良好时,用胰蛋白酶-EDTA消化液消化后进行后续慢病毒转染等实验。
1.2.5 慢病毒干扰特征基因SNRPA1的方法
在上海吉凯公司合成慢病毒序列(SNRPA1-RNAi,caTGGTATAATAGCCTTGTTT;阴性对照病毒CON313,TTCTCCGAACGTGTCACGT)。将慢病毒用新配置的完全培养基进行稀释,分装好的慢病毒存放于-80 ℃冰箱,避免反复冻融。将处于对数生长期的胃癌细胞AGS以特定密度(1.5×104)均匀接种到含有完全培养基的六孔板中,培养(37℃,5%CO2)24 h。更换新鲜的完全培养基。每孔加入20 μL感染增强液,放入恒温细胞培养箱中静置30 min。依次加入提前稀释的shRNA慢病毒和对照组病毒进行瞬时感染(置细胞培养箱中感染约12~16h)后换常规完全培养基。感染72 h后,用荧光显微镜观察细胞感染情况,当荧光细胞占细胞总数的70%~80%时,加入1 μL(2mg/μL)的嘌呤霉素,继续培养24 h。
1.2.6 实时定量RT-PCR测定方法
使用Trizol试剂及总RNA提取试剂盒提取胃癌细胞系总RNA,使用分光光度计测定RNA的浓度和纯度。在1%琼脂糖凝胶上进行RNA电泳,通过观察28S RNA及18S RNA来评估RNA的完整性。按照FastKing gDNA Dispelling RT SuperMix说明书将总RNA反转录成cDNA。由上海生工生物公司合成q-PCR特异性引物(GAPDH,上游序列TGACTTCAACAGCGACACCCA,下游序列CACCCTGTTGCTGTAGCCAAA;SNRPA1,上游序列AAAGTTCCGCAAGTCAGAGTAC,下游序列ACCAGCACCTGGATTAAAAGT)。根据SuperReal PreMix Color(SYBR Green)说明书配制反应溶液,使用实时荧光定量PCR仪进行PCR测定(反应条件:95℃,15min;95℃,10s;66℃,32s;共计40个循环)。实验重复3次。用GAPDH做内参,以2-△△Ct法计算基因相对表达量。
1.2.7 细胞增殖测定方法
计数2 000个处于对数生长期的AGS胃癌细胞接种于96孔细胞培养板中(设置空白组,加入同体积的完全培养基),每孔加入100 μL完全培养基。设置5个平行复孔,连续监测5 d。培养1 d(剩余四天的检测方法类似)后,将96孔细胞培养板从培养箱中取出,每个孔置换100 μL的新鲜完全培养基,再加入10 μL CCK-8到待测孔中,包好锡纸,在培养箱中继续孵育45 min。用酶标仪检测450 nm处的OD值。实验重复3次。
1.2.8 克隆形成方法
计数500个处于对数生长期的AGS细胞均匀接种到六孔板中,放入恒温细胞培养箱中培养7~10 d。每隔两天及时更换新鲜的完全培养基,为细胞提供充足的营养。在显微镜下观察到大部分细胞集落里的细胞数>50个时弃掉培养基,用无菌PBS清洗1次,每孔加入1 mL 4%多聚甲醛,置冰箱(4℃)固定细胞(60min)。再用PBS清洗1次,每孔加入1 mL吉姆萨染液于常温下染色(2min)。用ddH2O清洗细胞数次至无明显染色剂残留后晾干拍照,对显微镜下或肉眼可见的细胞集落进行计数。实验重复3次。
1.2.9 统计学方法
采用SPSS 26.0统计学软件处理数据。计量资料以均值±标准差表示。组间比较采用t检验,多组间比较采用单因素方差分析,两两比较采用LSD-t检验。以P<0.05为差异有统计学意义。
2 结果
2.1 高通量联合筛选结果
通过建立HCS技术,对前期获得的胃癌特征基因进行高通量差异基因增殖筛选分析,增殖倍数为实验组细胞数目增长倍数与shCtrl组细胞数目增长倍数的比值(细胞数目增长倍数为第五天细胞数目和第一天细胞数目的比值)。结果显示,每组在不同时间点的增殖差异有统计学意义(P<0.05),三组间的增殖差异有统计学意义(P<0.05),经两两比较发现,shCtrl与shPC、shSNRPA1的增殖差异有统计学意义(P<0.05)。在待检测的基因实验组中筛选增殖倍数≥2的实验组,找到特征基因SNRPA1,发现干扰SNRPA1基因后可以抑制胃癌细胞AGS增殖(FC=2.2,P<0.05)。(图1、表1-2)

图1 HCS高通量筛选结果图

表1 高通量差异基因增殖筛选结果

表2 SNRPA1高通量差异基因增殖筛选分析结果Table 2 Analysis results of HCS of SNRPA1
2.2 SNRPA1基因在胃癌中的表达水平、转录水平及分子相互作用机制
利用GEPIA2数据库分析特征基因SNRPA1在胃癌组织及正常组织中的表达情况,结果表明,胃癌组织中特征基因SNRPA1的表达水平明显高于正常组织(P<0.05)。(图2)

T(黑色)=胃癌组织,N(白色)=正常组织
通过Oncomine数据库比较了多种癌症和正常样本中特征基因SNRPA1在不同类型癌症中的转录水平,在与胃癌有关的数据集中发现SNRPA1的mRNA表达水平上调。另外,SNRPA1基因在11项肿瘤研究中呈现高表达,如膀胱癌、肺癌、乳腺癌等;在5项肿瘤研究中呈现低表达如脑癌、乳腺癌等。(图3)

图3 SNRPA1在不同类型癌症中的转录水平图
为进一步探索SNRPA1基因在胃癌发生发展中的作用及其相关分子间的相互作用机制,我们使用STRING数据库建立了与特征基因SNRPA1相关的蛋白相互作用网络,并选取可信度在0.9以上的基因,下载并导入CytoScape软件(mac版3.8.2)进行PPI网络分析。与特征基因SNRPA1相关的104个基因和SNRPA1基因组成3 634对网络关系(图4),平均节点度为69.2,PPI网络富集的P<1.0e-16。结果显示,包含SNRPA1在内的U2 snRNP相关蛋白参与了RNA加工、蛋白质复合体合成、核糖核蛋白体复合体合成、剪接体复合体合成、剪接体snRNP组装及RNA结合和核质运输等过程。

图4 PPI网络图
2.3 SNRPA1基因在6株胃癌细胞系中的表达
用qRT-PCR法分析6株胃癌细胞系中SNRPA1基因的表达情况(实验重复3次)并计算平均值(n=3)。确定△Ct为目的基因Ct值和内参基因Ct值的差值,△Ct值越大,目的基因表达丰度越低,△Ct值低于12时,说明该细胞中目的基因表达丰度较高。从定量PCR结果可以得出,特征基因SNRPA1在HGC27(6.74±0.39)、MKN-45(6.03±0.09)、MKN-74(5.26±0.36)、AGS(5.22±0.08)、NCI-N87(4.68±0.31)、KATO III(3.95±0.18)等细胞中的表达丰度较高(P<0.05)。
2.4 特征基因SNRPA1敲减效率(实时荧光定量PCR实验)
为了研究SNRPA1基因在胃癌发生、发展中的生物学作用,我们构建了干扰SNRPA1的慢病毒载体,用qRT-PCR法分析SNRPA1的基因转录情况来验证shRNA慢病毒对SNRPA1的干扰效率。以GAPDH为内源性对照,使用△Ct法定量每个样品中mRNA的相对量,来评估特征基因SNRPA1的基因表达情况。采用t检验分析各组间差异,以P<0.05表示差异具有统计学意义。结果显示,用shRNA慢病毒干扰特征基因SNRPA1后,抑制了胃癌细胞AGS在mRNA水平上的表达(P<0.05)。(表3)
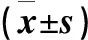
表3 SNRPA1 mRNA表达水平Table 3 mRNA expression level of
2.5 干扰SNRPA1基因对胃癌细胞AGS生长的影响(CCK-8法实验)
在胃癌细胞AGS中,我们分别转染SNRPA1干扰慢病毒shSNRPA1及阴性对照病毒,分析干扰特征基因SNRPA1对细胞生长的影响。采用t检验分析各组间差异,以P<0.05表示差异具有统计学意义。表4显示的是CCK-8检测干扰SNRPA1对胃癌细胞AGS细胞活力的影响,结果显示,每组细胞在不同时间点的活力不同,差异有统计学意义(P<0.05),在同一时间点,两组的细胞活力差异有统计学意义(P<0.05),与shCtrl组比较,shSNRPA1组可明显抑制胃癌细胞AGS的增殖(P<0.05)。
表4 干扰SNRPA1对胃癌细胞AGS细胞活力的影响(CCK-8实验)Table 4 Effects of interfering with SNRPA1 on the viability of gastric cancer cells AGS (CCK-8 experiment)
2.6 干扰SNRPA1基因对胃癌细胞AGS增殖的影响(克隆形成实验)
为了验证CCK-8实验结果,我们进一步采用集落形成实验证明干扰SNRPA1对胃癌细胞AGS生长的影响,研究转染SNRPA1干扰慢病毒shSNRPA1和阴性对照组慢病毒对AGS细胞集落形成能力的影响,将用慢病毒处理的AGS细胞接种于6孔板,培养7 d后观察形成的可见集落数量。采用t检验分析各组间差异,以P<0.05表示差异具有统计学意义。图5显示,经SNRPA1 shRNA慢病毒干扰后的胃癌细胞AGS形成的可见集落数量明显少于阴性对照病毒组。得出的数据结果证实了我们的观察,干扰特征基因SNRPA1后,可以显著抑制胃癌细胞AGS的集落形成能力(P<0.05)。(表5)
表5 干扰SNRPA1对胃癌细胞AGS细胞增殖的影响(克隆形成实验)Table 5 Effects of interfering with SNRPA1 on the proliferation of gastric cancer cells AGS cells
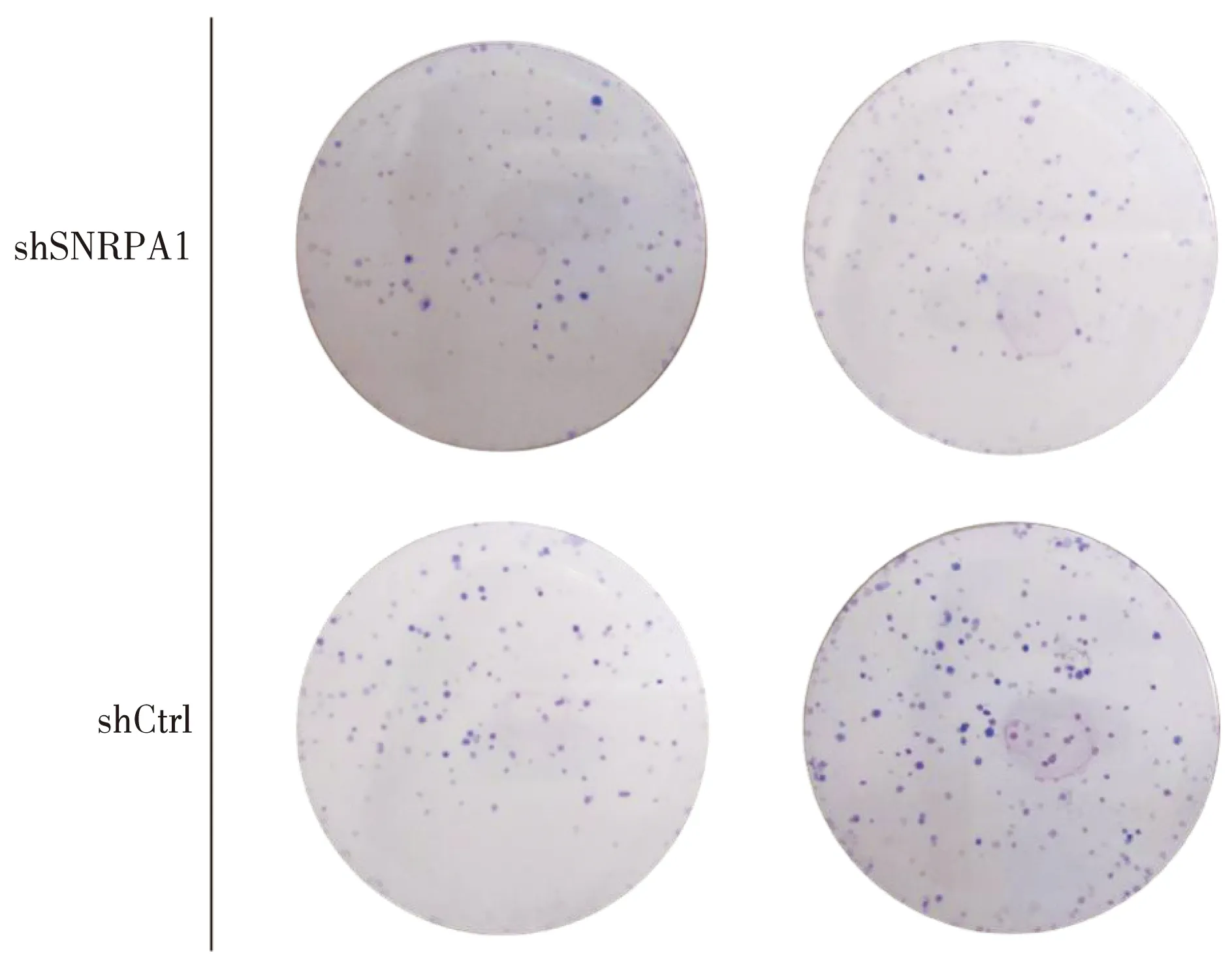
图5 可见集落图像
3 讨论
本研究结合TCGA分析胃癌差异表达谱发现SNRPA1在胃癌中高表达,进一步通过HCS技术发现干扰SNRPA1基因可以抑制胃癌细胞AGS的生长增殖。在对特征基因SNRPA1的表达水平进行差异性分析时发现,胃癌组织中SNRPA1基因的表达水平明显高于正常组织。特征基因SNRPA1在胃癌中的转录水平呈现高表达,在膀胱癌、乳腺癌、肺癌、胃癌、卵巢癌等组织中也发现SNRPA1基因高表达情况。已有学者发现,SNRPA1在多种肿瘤中高表达,在结、直肠癌中,其可通过调控磷脂酰肌醇-3-激酶调节亚基(phosphoinositide-3-kinase regulatory subunit 1,PIK3R1)和VEGFC、MKI67(marker of proliferation Ki-67)及周期蛋白依赖性激酶(cyclindependent kinases,CDK1)等基因的表达促进结、直肠癌细胞的增殖[2];在肝癌中,SNRPA1可以通过调控卵泡抑素样蛋白1(follistatin like protein 1,FSTL1)、碱性成纤维细胞生长因子(basic fibroblast growth factor,FGF2)和JAK2(Janus kinase 2,JAK2)、WNT5A及镁依赖性蛋白磷酸酶1A(protein phosphatase magnesium-dependent 1A,PPM1A)等基因的表达促进肝癌细胞的增殖[1]。还有学者发现[3],Wnt信号通路的转录因子TCF7L2、SNRPA1与rs386772267位点上的等位基因结合时,会提高胰腺癌的发病风险。
建立相互作用网络进行富集分析发现,与SNRPA1相关的大量基因参与了剪接体的转运通路,参与了RNA加工、mRNA(通过剪接体的)剪接和剪接体复合体合成及剪接体snRNP组装等生物学过程。目前的研究提示,剪接因子SF3家族与肿瘤的发生、发展密切相关,其中对SF3B的研究相对较多。作为SF3B的核心组件及癌症中最常见的突变剪接因子,SF3B1同时参与整个前体mRNA的剪接过程[4]。与正常组织相比,SF3B1在乳腺癌组织中过表达,并且与淋巴结转移有关,敲低SF3B1可以诱导乳腺癌细胞凋亡和细胞周期停滞从而降低癌细胞增殖、侵袭、迁移的能力,SF3B1突变能促进AKT、NF-kB信号输出,进而导致乳腺癌细胞的迁移和侵袭[5]。此外,SF3B1在肝癌[6]、骨髓增生异常综合征[7]、慢性淋巴细胞性白血病[8]、葡萄膜黑色素瘤[9]等肿瘤中均有突变发生。PHF5A作为参与肿瘤进展的关键剪接因子,在维持SF3B剪接体的稳定性方面起重要作用,PHF5A-SF3B复合物可调控细胞凋亡中的选择性剪接事件,敲低PHF5A可抑制乳腺癌细胞增殖、迁移[10]。PHF5A在肝癌中表达上调并可通过NF-κB信号促进肝癌细胞的侵袭和迁移[11]。总之,近年来发现了许多异常剪接与肿瘤发病相关,随着对剪接体结构与功能的不断解析,科学家们越来越关注剪接体相关蛋白的异常表达、基因异常剪接及转录后的调控与疾病的关系,并且开始关注靶向剪接。此前我们提出剪接体相关蛋白SNRPA1可能会影响胃癌细胞表达的假设,通过本研究我们证实了这一假设。本研究为探讨剪接体相关因子SNRPA1在胃癌发生、发展中的作用奠定了基础。
猜你喜欢
中国科学探险(2021年2期)2021-06-01 03:03:56
南方农业学报(2020年8期)2020-11-02 02:41:06
中央民族大学学报(自然科学版)(2016年3期)2016-06-27 07:55:28
兽医导刊(2016年12期)2016-05-17 03:51:54
生物学教学(2016年2期)2016-04-10 07:30:00
中国卫生标准管理(2015年3期)2016-01-14 03:41:46
中国医药生物技术(2015年4期)2015-12-26 08:26:36
科学大众(中学)(2015年11期)2015-12-09 09:06:51
医学研究杂志(2015年9期)2015-07-01 17:28:27
中国当代医药(2015年20期)2015-03-01 02:04:29
