
一种基于细胞测定维生素K环氧化物还原酶活性的方法
2022-06-22 02:30:48徐亚奇王一伊李飞邱稳辉沈滟沈国民
河南师范大学学报(自然科学版) 2022年4期
徐亚奇,王一伊,李飞,邱稳辉,沈滟,b,沈国民,b
(河南科技大学 a.基础医学院;b.河南省血栓与止血国际联合实验室,河南 洛阳 471023)
维生素K环氧化物还原酶(vitamin K epoxide reductase,VKOR)是位于内质网的整合膜蛋白,是维生素K循环中的限速酶,在血液凝集中发挥重要作用[1-2].在维生素K循环中,VKOR先后把维生素K环氧化物(vitamin K epoxide,VKO)转化为维生素K(vitamin K,VK)以及VK转化为二氢维生素K(vitamin K hydroquinone,VKH2)[3].VKH2作为辅因子参与γ-谷氨酰羧化酶(γ-glutamyl carboxylase,GGCX)对维生素K依赖性凝血因子进行γ羧基化修饰[4],这种翻译后修饰是维生素K依赖性凝血因子发挥生理功能的必要条件[5].VKOR是临床常用抗凝药华法林的特异性靶点[6].华法林通过抑制VKOR的活性,使VKO不能转化为VK和VKH2,从而阻断了维生素K循环,进而抑制维生素K依赖性凝血因子的γ羧基化修饰,发挥抗凝作用.在临床应用中,华法林存在个体用药量差异大和华法林耐受等问题[7-8],因此,开发克服华法林耐受的新型抗凝药具有重要应用价值和广泛应用前景.
为了鉴定化合物特异靶向抑制VKOR,需要利用VKOR活性实验进行测定.目前,VKOR活性实验方法有微粒体法[1-2]和报告基因法[9],其中微粒体法可以直接测定VKOR活性,但步骤较为繁琐.报告基因法虽然方便快捷,由于是间接测定VKOR活性,所以只能测定已知靶向VKOR化合物的抑制作用,而不能鉴定抑制VKOR的新型化合物.因此有必要建立一种方便快捷,且直接测定VKOR活性的方法.本研究利用高效液相色谱法建立了一种基于细胞测定VKOR活性的方法,该方法方便快捷,可用于鉴定靶向抑制VKOR的化合物.
1 材料与方法
1.1 材料
GGCX基因敲除细胞系293-GGCX-knockout为本实验室构建[10].DMEM培养基购自Hyclone公司,胎牛血清购自ABW公司,青霉素-链霉素溶液、胰蛋白酶溶液、TritonX-100购自北京索莱宝科技有限公司,色谱纯乙腈、正己烷和异丙醇购自天津科密欧化学试剂有限公司,维生素E醋酸酯(vitamin E acetate,VE)购自BBI公司,华法林、维生素K1(VK)、维生素K1-2,3-环氧化物(VKO)购自Sigma公司.
1.2 方法
1.2.1HPLC检测VK和VKO的条件
用高效液相色谱分析仪分析从细胞中萃取的维生素K,色谱柱为Eclipse XDB-C18(4.6 mm×150 mm,5.0 μm),流动相为乙腈-异丙醇(体积比为92.5∶7.5),流速1.2 mL/min,检测波长248 nm,柱温40℃,进样量30 μL,进样后洗脱时间为25 min.
1.2.2标准曲线样品、测试样品和内标样品的配制
首先配制500 μmol/L的VK溶液和50 μmol/L的VKO溶液.按倍比稀释法,用乙腈分别配制VK和VKO的标准工作液.VK标准工作液的浓度分别为500.00,250.00,125.00,62.50,31.20,15.60,7.80,3.90,1.96,0.98 μmol/L.VKO标准工作液的浓度分别为50.000,25.000,12.500,6.250,3.120,1.560,0.780,0.390,0.196,0.098 μmol/L.空白对照为乙腈溶液,用乙腈配制不同浓度的测试样品.维生素E醋酸酯为制备细胞样品的内标物,在萃取VK和VKO的过程中,用正己烷配制含维生素E醋酸酯的萃取试剂.
1.2.3细胞培养以及测定细胞内VKOR活性的预处理
293-GGCX-knockout细胞在体积分数为10%胎牛血清的DMEM完全培养基中培养.为了测定293-GGCX-knockout细胞的VKOR活性,把细胞铺至10 cm皿中,待细胞长至90%覆盖率时,加VKO至终浓度为10 μmol/L,继续培养4~6 h后收集细胞,萃取细胞内VK和VKO.对于华法林处理组,在加入VKO的同时,加入华法林,终浓度为10 μmol/L.
1.2.4VK和VKO的萃取与重悬
细胞处理后,弃培养液上清,收集细胞,用PBS洗3次,后收集细胞沉淀;加入1 mL细胞裂解液(50 mmol/L Tris-HCL pH7.5,150 mmol/L NaCL,体积分数1% TritonX-100),涡旋使细胞充分裂解,置于冰上20 min(期间再涡旋1次);向离心管中加入500 μL异丙醇,涡旋混匀30 s,再加入500 μL含维生素E醋酸酯(浓度为1 mmol/L)的正己烷,涡旋混匀30 s,封口膜封闭离心管,室温放置30 min(每隔10 min涡旋1次);2 000 r/min水平离心10 min,轻轻取出离心管(切勿震荡)置于管架上,此时溶液分为上层有机相和下层水相;吸取450 μL上层有机相于2 mL的棕色进样瓶中,将瓶开口避光室温放置使有机相自然挥发晾干(或氮气快速吹干).加入100 μL乙腈,使瓶壁及底部残留的脂溶性物质充分溶解,并转移到新的1.5 mL离心管中,13 000 r/min,4 ℃离心10 min,取上清进行HPLC分析.
2 结 果
2.1 方法的专属性
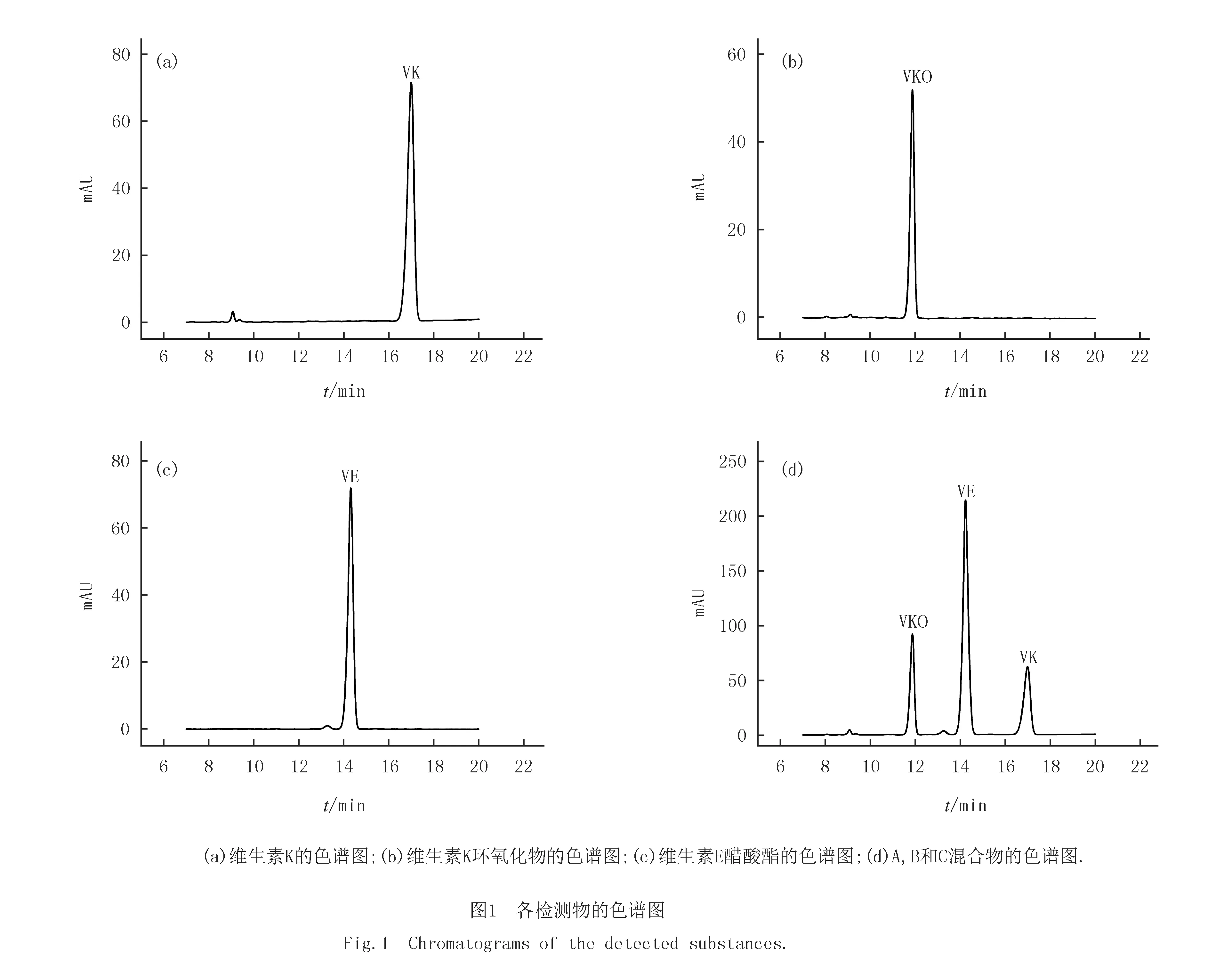
分别用乙腈配制不同浓度的含VK,VKO,VE和同时含有VK,VKO和维生素E醋酸酯的样品溶液,进行色谱分析检测,依据文献选取248 nm为检测波长[11].VK,VKO和维生素E醋酸酯的保留时间分别约为17.1,11.7和14.3 min(图1).三者峰形较好,在液相中分离良好,互不干扰.
2.2 标准曲线和定量限
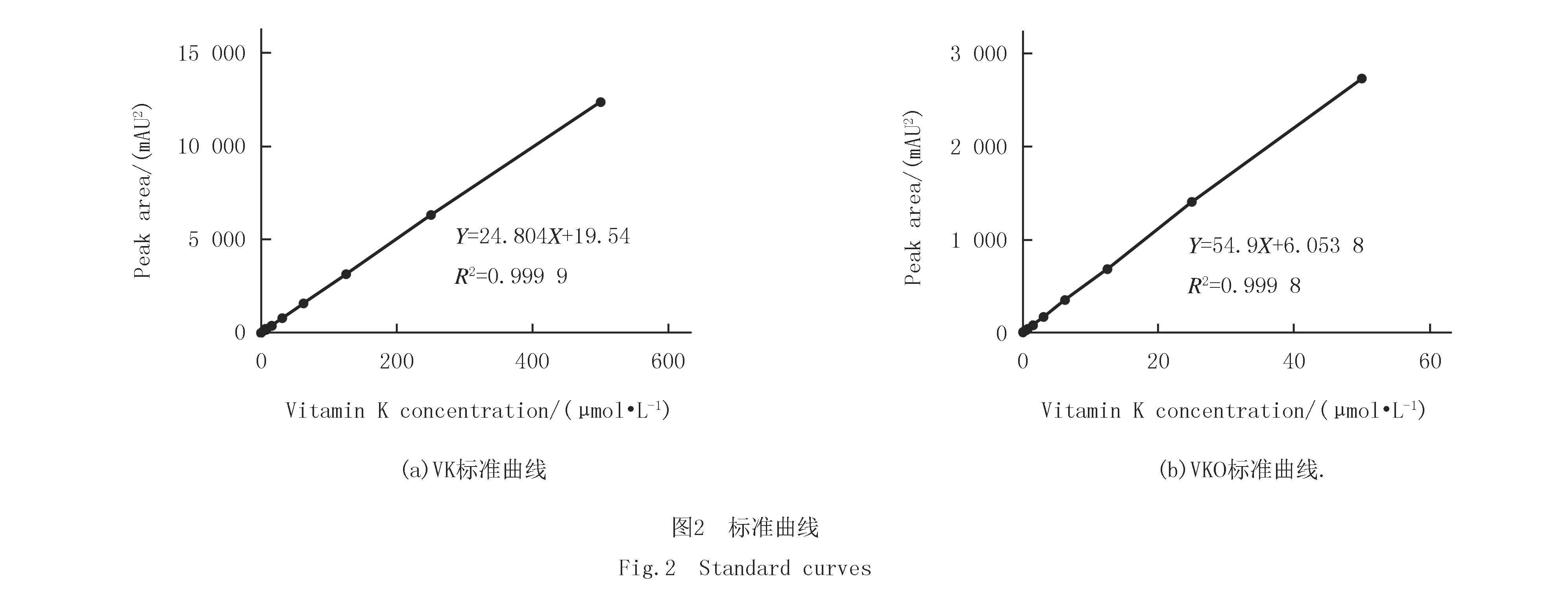
用HPLC分别分析VK和VKO的标准品,以标准品浓度为横坐标,对应的峰面积为纵坐标进行线性回归分析,得直线回归方程,即为标准曲线(图2).结果表明,VK在0.97~500.00 μmol/L的浓度范围内,其浓度与峰面积线性关系良好,回归方程为:Y=24.804X+19.54(图2(a),R2=0.999 9).VKO在0.097~50.000 μmol/L的浓度范围内,浓度与峰面积具有良好线性关系,回归方程为:Y=54.9X+6.05(图2(b),R2=0.999 8).因此,本研究中VK的定量限为0.97~500.00 μmol/L,VKO的定量限为0.097~50.000 μmol/L.
2.3 精密度和稳定性
为了检测方法的精密度和回收率,分别配制低、中、高(62.5,125.0,375.0 μmol/L)3种浓度的VK质控样品和低、中、高(6.25,12.5,37.5 μmol/L)3种浓度的VKO质控样品.将上述质控样品置于-20 ℃冰箱中保存,分别在第1、2、3 d进行色谱分析,每个质控样品每天测定5次,依据上述标准曲线,分析日内精密度、日间精密度和回收率(表1).结果显示各浓度VK的日内精密度为0.1%~1.2%,日间精密度为0.2%~1.6%,回收率为94.0%~98.9%;各浓度VKO的日内精密度为0.30%~1.70%,日间精密度为0.9%~3.2%,回收率为94.3%~103.2%,均符合要求(表1).
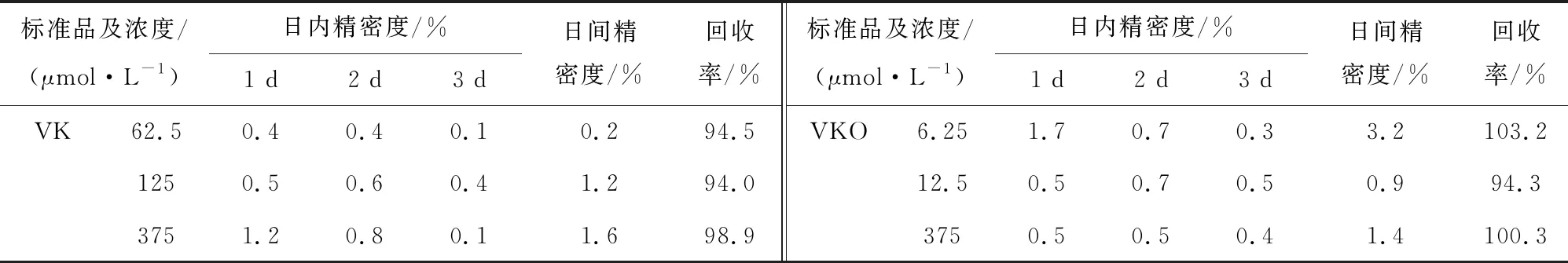
表1 VK和VKO的精密度和回收率结果
2.4 GGCX基因敲除细胞中测定VKOR活性
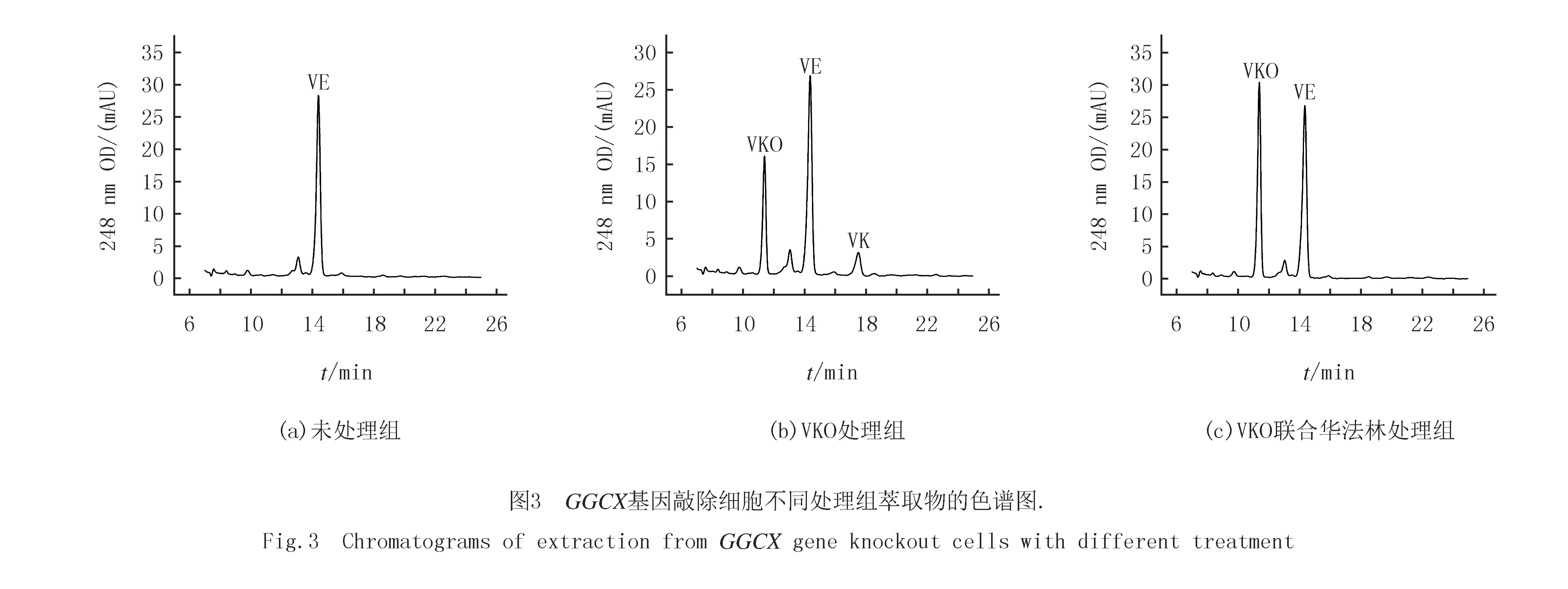
收集不同处理组的GGCX基因敲除细胞,按1.2.4方法萃取VK和VKO,在萃取过程中加入维生素E醋酸酯为内标,用于分析不同样品在萃取过程中的误差.结果表明,在未处理组,可以检测到特异的维生素E醋酸酯的峰,且峰值与VKO处理组相似,但没有检测到VK和VKO的特异峰(图3(a)),说明细胞内源的VK和VKO极少,检测不到,因此用该细胞测定VKOR活性时,内源的VK和VKO对测定VKOR活性的影响可以忽略.在VKO处理组,可以检测到VK和VKO的峰,说明细胞吸收VKO后,在VKOR的催化下部分VKO被还原为K(图3(b));依据峰面积和标准曲线分别计算出VK和VKO的浓度,VKO的转化率为CVK/(CVK+CVKO),可计算出VKO的转化率约为35.5%.在VKO与华法林共处理组(图3(c)),由于华法林特异抑制VKOR活性,导致VKO不能被VKOR还原为K,所以在色谱图上只检测到VKO,而没有检测到VK(图3(c)),因此VKO的转化率为0.
3 讨 论
VKOR作为开发抗凝药的重要靶点,其活性测定对鉴定新型VKOR抑制剂具有重要的应用价值[12],但由于该蛋白是一种整合膜蛋白,其活性测定方法较为有限.目前,测定VKOR活性的常用方法为基于报告基因的方法,是一种间接测定VKOR活性的方法,不能用于鉴定靶向抑制VKOR的新型抑制剂,因为该方法还可以分析维生素K循环中GGCX的活性[12].本研究建立了一种基于HPLC检测VK和VKO的方法,该方法简便快速,准确性高,重复性好,且各检测物色谱峰分离良好.在流动相优化过程中,乙腈和异丙醇的比例对峰形和出峰时间很重要,减少异丙醇比例,各检测物的出峰时间将延长,且峰形可能为非对称单峰.为了兼顾各检测物的分离效果和缩短每个样品的检测时间,流动相乙腈和异丙醇的体积比例为92.5∶7.5.另一方面,由于维生素K以3种形式在细胞内循环,在测定VKOR活性中,为使VKOR还原的VK不被GGCX转化为KO,本研究选择在GGCX基因敲除的细胞中测定VKOR活性,可以排除细胞中GGCX活性对VKOR活性测定的干扰.在从细胞中萃取VK和VKO的过程中,为了分析萃取操作过程对结果的影响,实验选择维生素E醋酸酯为内标,可分析操作过程对结果的影响[12].综上,本研究通过建立检测VK和VKO的方法,利用GGCX基因敲除的细胞直接测定VKOR活性,将为靶向VKOR抑制剂的鉴定和新型抗凝药的研发提供技术支持.
猜你喜欢
煤化工(2022年3期)2022-07-08 07:24:42
保健医苑(2022年4期)2022-05-05 06:11:06
World Journal of Clinical Cases(2020年17期)2020-09-18 08:03:24
检验医学与临床(2020年1期)2020-01-10 04:44:22
中华老年多器官疾病杂志(2016年9期)2016-04-28 08:52:46
中西医结合心脑血管病杂志(2016年20期)2016-03-01 04:20:34
中国资源综合利用(2016年10期)2016-01-22 08:36:09
应用海洋学学报(2015年2期)2015-11-22 07:36:40
现代检验医学杂志(2015年6期)2015-02-06 01:44:25
中国医学科学院学报(2012年3期)2012-03-25 13:59:02
