
基于转录组测序的棘胸蛙SSR和SNP分子标记开发
2022-06-15 11:37:33魏朝宇谢永广魏秀英罗华辉陈敦学
南方农业学报 2022年3期
关键词:分布特征
魏朝宇 谢永广 魏秀英 罗华辉 陈敦学


摘要:【目的】基于轉录组开发SSR和SNP分子标记用于评价棘胸蛙(Quasipaa spinosa)遗传多样性,为其种质资源的创新利用提供理论支撑。【方法】采用TRIzol试剂盒提取棘胸蛙肝脏、肌肉和肾脏组织总RNA,构建cDNA文库后利用Illumina HiSeq 2500测序平台进行高通量测序,通过MISA对棘胸蛙转录组测序数据进行SSR检索,并以SAMtools和VarScan v.2.2.7进行SNP查找。【结果】棘胸蛙转录组测序共获得93887条非冗余基因(Unigenes),序列总长度达91352712 bp,且所有转录组Q30均超过95.00%。在93887条Unigenes中发现33019个SSRs,其中21966条Unigenes含有SSR,6688条Unigenes含有超过1个SSR;以单核苷酸重复型SSR数最多,达25788个,且出现频率最高(27.47%)。SSR的平均长度以四核苷酸重复型最长,达35.47 bp;棘胸蛙SSR以(A/T)n为绝对优势重复基元,占总SSR的65.51%,然后依次为(C/G)n、(AT/AT)n、(AC/GT)n、(AG/CT)n、(AAT/ATT)n、(AGG/CCT)n,分别占总SSR的12.59%、5.66%、5.55%、3.31%、1.55%和1.30%。在33019个SSRs中,核苷酸重复次数主要集中在5~25次,占总SSR的99.91%,且大部分SSR位于非编码区,仅有1633个SSRs位于编码区;长度≥12 bp的SSR共计17244个,占总SSR的58.53%。挑选120对SSR引物进行引物有效性验证,发现有57对引物扩增出单一条带,且条带大小与预期结果一致。对棘胸蛙转录组序列进行SNP检索,共发现87634个SNPs,其中,56300个SNPs属于转换位点、31334个SNPs属于颠换位点,转化/颠换比达1.80,碱基的转换频率明显高于颠换频率。【结论】利用高通量转录组测序开发棘胸蛙SSR和SNP分子标记是一种切实可行的方法,能开发出通用性较高、数量较多、覆盖性较广的分子标记。棘胸蛙具有中度偏高的遗传多样性,可作为种质材料进一步开发利用。
关键词:棘胸蛙;SSR分子标记;SNP分子标记;分布特征;转录组测序
中图分类号: S917;S966.39 文献标志码: A 文章编号:2095-1191(2022)03-0759-09
SSR and SNP molecular marker development based on Quasipaa spinosa transcriptome sequencing
WEI Zhao-yu, XIE Yong-guang, WEI Xiu-ying, LUO Hua-hui, CHEN Dun-xue
(College of Animal Science, Guizhou University/Key Laboratory of Animal Genetics and Breeding and Reproduction of Plateau and Mountain Animals/Research Center of Fishery Resources and Environment, Guizhou University,
Guiyang, Guizhou 550025, China)
Abstract:【Objective】To investigate the characteristics of SSR and SNP locus based on the transcriptome for evalua-ting the genetic diversity of Quasipaa spinosa, so as to provide appropriate molecular markers for the innovative ways of germplasm resource application. 【Method】Total RNA was extracted from liver, muscle and kidney tissues of Q. spinosa to build a TRlzol kit. cDNA library and then the library was high-throughput sequenced by the Illumina HiSeq 2500 sequencing platform. Microsatellite searching software MISA was used to screen and analyze microsatellite (SSR) in the Q. spinosa transcriptome while the software SAMtools and VarScan v.2.2.7 were used for searching SNP loci. 【Result】 93887 non-redundant unigenes with a total sequence length of 91352712 bp were obtained from the transcriptome sequencing of Q. spinosa, and all transcriptome Q30 were over 95.00%. Among the 93887 unigenes, 33019 potential SSR markers were identified and 21966 unigenes contained SSR loci. In additional, a total of 6688 unigenes had more than one SSR locus. The dinucleotide was the highest at 25788 and the frequency of occurrence frequency was the highest at 27.47%. The average length of SSR was 35.47 bp. (A/T)n was the absolutely dominant repeat motif of Q. spinosa SSR, accounting for 65.51% of the total SSRs, followed by (C/G)n, (AT/AT)n, (AC/GT)n, (AG/CT)n, (AAT/ATT)n, and accounting for 12.59%, 5.66%, 5.55%、3.31%, 1.55% and 1.30% of the total SSRs, respectively. Among the 33019 potential SSR markers, the times of repetition was mainly between 5-25 times, accounting for 99.91% of the all SSRs. In additional, only 1633 located in the coding area. 17244 SSR loci whose length≥12 bp accounted for 58.53% of the total SSR loci. 120 pairs of SSR primers were selected to verify the validity of the primers and 57 pairs of primers amplified a single band, and the band size was as expected. 87634 SNPs were identified (56300 transitions and 31334 transversions) from mapping sequencing reads to assembled unigenes, the transition/transversion ratio was approximately1.80 and the frequency of base transition is higher than that of transversion. 【Conclusion】High-throughput transcriptome sequencing is a feasible method to develop SSR and SNP molecular markers, which can develop molecular markers with universality, large number and wide coverage. Q. spinosa has a moderately high genetic diversity, so it can be used as germplasm materials for further development and utilization.8C9BE7E2-0628-425C-8B6F-560A759F3A42
Key words:Quasipaa spinosa; SSR molecular marker; SNP molecular marker; distribution characteristics; transcriptome sequencing
Foundation items: Guizhou Science and Technology Plan Program (QKHZC〔2019〕2344)
0 引言
【研究意义】分子标记开发是基因组学与分子生物学应用于生产实践的重要手段,筛选出合适的分子标记进行遗传多样性分析或良种选育能有效推动种质资源开发与创新利用(Gao et al.,2012),目前使用的分子标记主要有微卫星(SSR)分子标记、表达序列标签(EST)分子标记及单核苷酸多态性(SNP)分子标记等。SSR又称简单重复序列,主要由高突变的核心序列和保守的侧翼序列组成,具有数量多、分布广泛且均匀、杂合率高、重复性好且数据易统计等优点(Wang et al.,2015)。SNP包含转换(C/T和G/A)和颠换(C/G、C/A、T/A和T/G)2种类型,具有高密度、可扩展性和全基因组分布的特征(Trick et al.,2009),尤其是编码区的SNP位点可能对蛋白功能和基因表达产生极大影响,已广泛应用于动物遗传育种(Garrido-Cardenas et al.,2018)。因此,开发大量分子标记对物种种质资源保护与创新利用起到积极的推动作用。【前人研究进展】随着转录组测序技术的逐渐成熟及其成本的不断降低,利用高通量测序一次性获得大量的SSR和SNP位点已成为现实。在水产动物研究中,Gao等(2012)通过转录组测序同时筛选获得团头鲂(Megalobrama amblycephala)的SSR和SNP位点;Li等(2015)通过高通量测序挖掘出大鳞副泥鳅(Paramisgurnus dabryanus)的15106个潜在SSR位点;Zhao等(2019)通过比较野生和人工养殖白鲈(Morone chrysops)转录组数据,获得13872个潜在SSR位点,并最终筛选得到57个差异位点;Yá?ez等(2020)通过基因组开发出尼罗罗非鱼(Oreochromis niloticus)的5000个高质量SNP位点,并利用SNP位点对尼罗罗非鱼进行群体分类。此外,许多水产动物的SSR和SNP位点均通过高通量测序技术被批量开发,如虾夷扇贝(Patinopecten yessoensis)(倪守胜等,2018)、宽体金线蛭(Whitmania pigra Whitman)(熊良伟等,2018)、红鳍东方鲀(Takifugu rubripes)(Pandey et al.,2018)、翘嘴鳜(Siniperca chuatsi)(孙海林等,2019)、波纹唇鱼(Cheilinus undulatus)(刘洪涛等,2020)、中华绒螯蟹(Eriocheir sinensis)(徐杰杰等,2021)及石斑鱼(Epinephelus tukula)(Hsu et al.,2021)等。至今,已发现的两栖类大概有7200种,具有较高的基因复杂性(Mable et al.,2011),且两栖类研究主要集中在热带爪蟾(Silurana tropicalis)(Hellsten et al.,2010)和非洲爪蟾(Xenopus laevis)(Borodinsky,2017)。显然,这2种模式生物无法代表所有两栖类动物,因此迫切需要丰富两栖类基因组序列,为两栖类的资源保护与创新利用提供数据支撑(Savage et al.,2014)。【本研究切入点】棘胸蛙(Quasipaa spinosa)隶属于脊索动物门(Phylum Chordata)两栖纲(Amphibia)蛙科(Ranidae)蛙属(Rana),为大型食用蛙类,主要分布在我国贵州及江西等地区(Hu et al.,2017),以及少量分布在越南,具有高蛋白、低脂肪和高不饱和脂肪酸的特点。从20世纪80年代起我国开始对棘胸蛙进行人工养殖(Chan et al.,2014),但由于养殖过程中相互引种,导致目前很多养殖场养殖的棘胸蛙并非纯种(Ye et al.,2013),因此亟待对棘胸蛙遗传背景进行摸底研究,为其人工选种和资源多样性分析提供参考依据。【拟解决的关键问题】利用转录组数据筛选SSR和SNP位点,分析其核苷酸重复类型及序列分布特征,并利用所筛选的SSR引物在5个棘胸蛙养殖群体中进行SSR位点验证,开发相关分子标记用于评价棘胸蛙遗传多样性,为其种质资源的创新利用提供理论支撑。
1 材料与方法
1. 1 试验材料
供试棘胸蛙由贵州省遵义市播州区洪关苗乡石蛙养殖专业合作社提供,共9尾,不存在引种行为,通过形态鉴定为纯种棘胸蛙。经MS-222麻醉后进行解剖处理,取其肝脏、肌肉和肾脏3个组织,按3尾一组进行随机组合和混样,共计9个样品,液氮保存备用。
1. 2 RNA提取与cDNA文库构建
采用TRIzol试剂盒提取组织总RNA,利用NanoDrop 2000进行RNA浓度和纯度检测,1.5%琼脂糖凝胶电泳进行完整性检测,Agilent 2100测定RIN值。通过带有Oligo(dT)的磁珠从总RNA中分离出mRNA,加入Fragmentation Buffer将mRNA随机断裂成短片段后,以mRNA为模板利用随机引物反转合成cDNA第一链,在此基础上再合成cDNA第二链,形成稳定的双链结构;随后加ploy(A)尾巴并连接Adaptor,对连接Adapter的产物进行纯化和片段分选,进行PCR擴增,纯化得到最终的cDNA文库。cDNA文库质量检测合格后,委托上海美吉生物医药科技有限公司利用Illumina HiSeq 2500测序平台进行高通量测序。8C9BE7E2-0628-425C-8B6F-560A759F3A42
1. 3 数据分析
利用MISA(https://webblast.ipk-gatersleben.de/misa/)进行SSR检索,设SSR的筛选条件为单核苷酸重复次数≥10,二核苷酸重复次数≥6,三、四、五、六核苷酸重复次数≥5。参照Li和Dewey(2011)的方法,利用SAMtools和VarScan v.2.2.7进行SNP查找。
2 结果与分析
2. 1 棘胸蛙转录组测序及SSR检索结果
通过对棘胸蛙肝脏、肌肉和肾脏组织进行转录组测序,并进行序列拼接和组装,共获得93887条非冗余基因(Unigenes),序列总长度达91352712 bp,且所有转录组Q30均超过95.00%,即获得较好的测序结果。在获得的93887条Unigenes中最长序列达50351 bp,N50为1434 bp,平均长度为973 bp。在93887条Unigenes中共发现33019个SSRs,其中21966条Unigenes含有SSR,6688条Unigenes含有超过1个SSR,3560条Unigenes含有复杂的SSR(表1)。
2. 2 棘胸蛙SSR重复类型分析结果
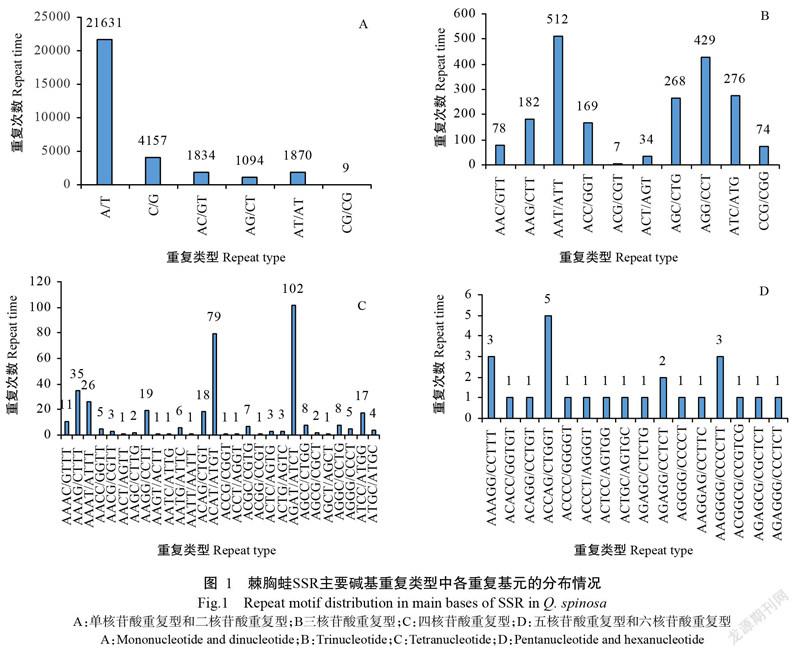
将检索获得的33019个SSRs按重复类型进行分类,可分为单核苷酸重复、二核苷酸重复、三核苷酸重复、四核苷酸重复、五核苷酸重复和六核苷酸重复6种类型,其中以单核苷酸重复型SSR数最多,达25788个,且出现频率最高(27.47%),然后依次是二核苷酸重复~六核苷酸重复型SSR,出现频率分别为5.12%、2.16%、0.40%、0.02%和0.01%(表2)。SSR的平均长度以四核苷酸重复型最长,达35.47 bp,然后依次为六核苷酸重复型(33.00 bp)、五核苷酸重复型(25.83 bp)、二核苷酸重复型(18.57 bp)、三核苷酸重复型(17.41 bp)和单核苷酸重复型(12.19 bp)。
在所有的SSR中共发现60种重复基元类别,以四核苷酸重复基元最多,达28种,然后依次为五核苷酸重复基元11种、三核苷酸重复基元10种、六核苷酸重复基元5种、二核苷酸重复基元4种、单核苷酸重复基元2种(图1)。在四核苷酸重复的28个基元类别中,数量和分布特征差异明显,SSR的优势重复基元为AGAT/ATCT,占四核苷酸重复基元的27.28%(表2),而AACT/AGTT、AAGT/ACTT、AATC/ATTG、AATT/AATT、ACCG/CGGT、ACCT/AGGT、ACGG/CCGT和AGCT/AGCT等重复基元仅出现1次;五核苷酸和六核苷酸重复的基元分布较均匀,单核苷酸重复中则以A/T为优势重复基元,占单核苷酸重复基元的83.88%;二核苷酸重复基元中除CG/CG仅重复出现9次外,其余3种基元分布较均匀[AC/GT(1834次),AG/CT(1094次),AT/AT(1870次)],以AC/GT为优势重复基元(38.15%);三核苷酸重复基元分布相对不均衡,以AAT/ATT为优势重复基元(25.23%),共重复出现512次,然后依次是AGG/CCT(429次)、ATC/ATG(276次)、AGC/CTG(268次)、AAG/CTT(182次)、ACC/GGT(169次)、AAC/GTT(78次)、CCG/CGG(74次)和ACT/AGT(34次),重复次数最少的基元为ACG/CGT,仅出现7次。整体而言,棘胸蛙SSR以(A/T)n为绝对优势重复基元,占总SSR的65.51%,然后依次为(C/G)n、(AT/AT)n、(AC/GT)n、(AG/CT)n、(AAT/ATT)n、(AGG/CCT)n,分别占总SSR的12.59%、5.66%、5.55%、3.31%、1.55%和1.30%。
2. 3 棘胸蛙SSR位点在Unigenes中的分布特征
将33019个SSRs在93887条Unigenes中进行搜索及比对分析,结果(图2)发现大部分SSR均位于不确定区域(19075个),其次位于3'端(6719个)和5'端(2032个),在编码区仅发现1633个SSRs。在1633个SSRs中,以单核苷酸基重复和三核苷酸重复为主,分别为679和630个;在编码区还出现部分复合型SSR(135个)。
2. 4 棘胸蛙SSR重复次数分布情况
SSR重复次数与重复片段长度也存在一定的分布特征,经统计发现棘胸蛙SSR的核苷酸重复次数分布范围主要在5~75次,跨度较大,其中重复次數集中在5~25次的SSR占总SSR的99.91%,其重复次数的具体分布情况见表3。
2. 5 棘胸蛙SSR长度分布情况
棘胸蛙转录组中SSR长度统计结果显示,所有SSR总长度为569846 bp,占转录组序列全长的0.62%。棘胸蛙SSR长度存在明显差异,长度最短的为10 bp,最长的≥100 bp。整体来看,SSR长度主要集中在10~14 bp(占68.07%),且SSR的数量随着序列长度的增加急剧下降,分布在15~19 bp、20~29 bp、30~49 bp、10~100 bp、>100 bp的SSR分别为4723个(占16.03%)、1950个(占6.62%)、1226个(占4.16%)和1287个(占4.37%),只有2.28%的SSR长度超过100 bp(图3)。棘胸蛙SSR长度在20 bp以上的仅有5133个,占总SSR的17.42%;而长度介于12~20 bp的SSR为12111个,占总SSR的41.11%;长度≥20 bp的位点共计17244个,占总SSR的58.53%,说明棘胸蛙具有中度偏高的片段长度多态性。8C9BE7E2-0628-425C-8B6F-560A759F3A42
2. 6 棘胸蛙SSR的初步验证结果
按去除单核苷酸重复和复杂重复类型的原则,选择SSR两端序列长度≥50 bp的序列进行引物设计,随机挑选序列并设计500对SSR引物,挑选其中的120对SSR引物进行引物有效性验证。以5个棘胸蛙养殖群体的基因组DNA为模板进行PCR扩增和引物筛选,发现120对SSR引物中共有57对引物扩增出单一条带,且条带大小与预期结果一致。表4为部分SSR引物在棘胸蛙个体中的扩增多态性。
2. 7 棘胸蛙转录组中SNP的特征分析结果
对棘胸蛙转录组序列进行SNP检索,共发现87634个SNPs(图4),发生频率为1042 bp序列会出现1个SNP。在搜索获得的87634个SNPs中,有56300个SNPs属于转换位点、31334个SNPs属于颠换位点,转换/颠换比达1.80。以A/G转换的发生频率最高,占SNP总数的32.26%,其次是C/T转换,占SNP总数的31.98%,其余4种颠换类型(A/T、A/C、T/G和C/G)分别占SNP总数的8.1%、8.84%、8.40%和10.39%。2种高变异类型(A/G和C/T)均属于转换类型,而剩下的4种颠换类型所占比例均低于15.00%,即碱基的转换频率明显高于颠换频率。
3 讨论
3. 1 测序深度与筛选方法对SSR特征分析的影响
随着测序技术的成熟及其成本的降低,越来越多研究通过高通量测序获得水产动物的SSR位点(Zhang et al.,2014;Li et al.,2015;Pandey et al.,2018),但测序深度对SSR的开发具有重要影响。Pandey等(2018)通过红鳍东方鲀的全基因组序列开发出139057个潜在SSRs,但孙赛红(2014)通过EST序列查找仅发现27914个SSRs;梁霞等(2021)通过鲤鱼(Cyprinus carpio)全基因组测序共获得837004个完整SSRs,而岳华梅等(2016)通过转录组测序仅获得13652个SSRs;迟天舒(2020)对东北林蛙的基因组测序发现792550个潜在SSRs,而在转录组序列中仅发现30830个潜在SSRs,在同属于两栖类的四川湍蛙和棘腹蛙研究上也存在类似现象(Xia et al.,2018)。可见,SSR的开发与测序深度密切相关。本研究通过对棘胸蛙肌肉、肝脏和肾脏3个组织的9个样品进行转录组测序,所得序列总长度达91352712 bp,且所有转录组Q30均超过95.00%,筛选获得33019个SSRs,具有较好的测序深度,说明基于转录组测序开发获得的SSR具有较好的覆盖性和可信度。此外,SSR分析设置重复参数不一样,也会影响到SSR的开发。熊良伟等(2018)进行宽体金线蛭SSR位点开发时设置为单核苷酸重复≥13,二核苷酸重复≥6;孙海林等(2019)进行翘嘴鳜SSR开发时设单核苷酸重复≥13次,二核苷酸重复≥7次;而李超等(2015)在开发牙鲆(Paralichthys olivaceus)SSR位点、倪守胜等(2018)开发虾夷扇贝SSR位点时并未统计单核苷酸重复。不同的统计方法必然导致最终统计的SSR数量有所差异。本研究则按照默认设为单核苷酸重复≥次数10,二核苷酸重复次数≥6。
3. 2 棘胸蛙SSR的重复类型与分布特征
不同物种的SSR在分布和丰度上存在明显差异(黄杰等,2012)。本研究中,棘胸蛙以单核苷酸重复为主,达25788个,出现频率为27.47%,且以A/T为主(83.88%),但由于单核苷酸重复存在一些poly(A)或假基因,而导致统计结果出现偏差,故在讨论中暂不考虑单核苷酸重复。已有研究证实,SSR数量随着重复长度的增加而减少(李清莹等,2019),进化较高的物种通常存在较多的低级重复单元(Harr and Schl?tterer,2000;刘洪涛等,2020)。在棘胸蛙中(不考虑单核苷酸)以二核苷酸重复为主(4807个),占除去单核苷酸重复后所有SSR的66.48%。其次为三核苷酸重复,四核苷酸重复、五核苷酸重复和六核苷酸重复出现的次数较少。在中国大鲵(Andrias davidianus)的研究中也是以单核苷酸重复为主,然后依次为二核苷酸重复和三核苷酸重复(Huang et al.,2017),且出现随SSR长度增加其数量减少的现象,可能与序列的稳定性和进化压力有关(Jo et al.,2021)。在大鳞副泥鳅(Li et al.,2015)、银鲳(Pampus argenteus)(刘磊等,2016)、兴国红鲤(C. carpio var)(岳华梅等,2016)及波纹唇鱼(刘洪涛等,2020)等鱼类中也发现类似的现象,但存在种属差异性(马秋月等,2013),在同为两栖类的中国小鲵(Hynobius chinensis)中则以三核苷酸重复出现频率最高(Che et al.,2014),且水产动物中广泛存在类似现象(Zhang et al.,2008;Bai et al.,2009;倪守胜等,2018)。
进一步分析棘胸蛙SSR的优势重复基元,发现二核苷酸重复中以AC/GT为优势重复基元,而GC/CG重复的SSR较少,仅重复出现9次。中国大鲵的SSR研究发现,也是以GT/AC重复基元为主(Huang et al.,2017),类似结果在其他水生动物中也有发现,包括牙鲆(李超等,2015)、銀鲳(刘磊等,2016)及中华绒螯蟹(徐杰杰等,2021)等,但在中国大鲵(Huang et al.,2017)、波纹唇鱼(刘洪涛等,2020)和日本沼虾(Macrobrachium nipponense)(赵燕等,2020)中未发现GC/CG重复基元。究其原因可能是C≡G间氢键较稳固,在DNA复制过程中不易产生滑移,因此SSR位点较少(Zhao et al.,2011)。此外,SSR在基因序列中的分布特征存在不均一性,分布在基因编码区的较少(Rhode and Roodt-Wilding,2011),而开发位于编码区的SSR位点对开展SSR跨种通用性研究具有重要意义(Wang et al.,2007)。在本研究中,在编码区仅发现1633个SSRs(占4.95%),且主要以单核苷酸重复和三核苷酸重复为主,位于编码区的三核苷酸重复SSR对基因转录翻译的影响较小,不仅保证其遗传的稳定性,还能增加变异和进化效率。类似结果在其他鱼类中也有发现,波纹唇鱼中仅有1773个SSRs位于编码区,占7.9%(刘洪涛等,2020);在黑鲷(Acanthopagrus schlegelii)和真鲷(Pagrus major)中SSR在编码区的发生频率也较较低,仅为8.5%和7.8%(曹广勇等,2019)。8C9BE7E2-0628-425C-8B6F-560A759F3A42
3. 3 棘胸蛙的遗传多态性
SSR和SNP均是检验物种多态性的重要分子标记,而影响SSR多态性的重要因素为核苷酸重复序列长度,多态性较高的SSR长度一般≥20 bp,多态性中等的SSR长度一般在12~20 bp(杨芩等,2021)。棘胸蛙的SSR长度主要集中在12~14 bp,其中SSR长度大于12 bp的序列有17244 bp,占总SSR的58.53%,即棘胸蛙具有中度偏高的多态性,与Zheng等(2009)通过线粒体序列(12S和16S)进行棘胸蛙多态性分析的结果存在差异;Yu等(2016)通过10个SSR位点和线粒体CYTB序列也证明棘胸蛙具有较高的遗传多样性。这可能是由于本研究选用的棘胸蛙养殖群体经过多年自繁自养后,近亲繁殖较严重,其资源退化、遗传多样性有所降低所致。
基于SNP进行多态性分析,发现棘胸蛙转录组中的转化/颠换比达1.80,类似结果在不少鱼类中也有发现。波纹唇鱼中的转化/颠换比为1.95(刘洪涛等,2020),石斑鱼中的转化/颠换比达2.52(Hsu et al.,2021),可能與不同物种生物在进化过程中承受的选择压力不同有关(Zhang et al.,2018)。此外,棘胸蛙转录组中SNP的出现频率远高于SSR,说明单核苷酸变异可能更易发生(刘洪涛等,2020)。2种高变异类型(A/G和C/T)均属于转换类型,C/T含量高可能是由于胞嘧啶(C)易发生脱氨基作用转变成胸腺嘧啶(T),引起C/T变异发生频率高,而A/G含量高与快速生长相关(Tsai et al.,2015;Hsu et al.,2021)。
4 结论
利用高通量转录组测序开发棘胸蛙SSR和SNP分子标记是一种切实可行的方法,能开发出通用性较高、数量较多、覆盖性较广的分子标记。棘胸蛙具有中度偏高的遗传多样性,可作为种质材料进一步开发利用。
参考文献:
曹广勇,张志勇,张志伟,陈淑吟,祝斐,贾超峰,陈自强,曾海峰,汤晓建. 2019. 黑鲷、真鲷及其杂交子代基因编码区微卫星序列及密码子偏好性分析[J]. 海洋与湖沼,50(5):1108-1115. [Cao G Y,Zhang Z Y,Zhang Z W,Chen S Y,Zhu F,Jia C F,Chen Z Q,Zeng H F,Tang X J. 2019. Analysis of the microsatellite sequences and codon bias of the coding sequence in Acanthopagrus schlegelii,Pagrus major and their hybrid progenies[J]. Oceanologia et Limnologia Sinica,50(5):1108-1115.] doi:10.11693/hyhz20190200038.
迟天舒. 2020. 基于高通量测序东北林蛙微卫星标记开发与应用[D]. 沈阳:沈阳师范大学. [Chi T S. 2020. Deve-lopment and application of microsatellite markers based on next-generation sequencing for Rana dybowskii[D]. Shenyang:Shenyang Nomal University.] doi:10.27328/d.cnki.gshsc.2020.000539.
黄杰,杜联明,李玉芝,李午佼,张修月,岳碧松. 2012. 红原鸡全基因组中微卫星分布规律研究[J]. 四川动物,31(3):358-363. [Huang J,Du L M,Li Y Z,Li W J,Zhang X Y,Yue B S. 2012. Distribution regularities of microsatellites in the Gallus gallus genome[J]. Sichuan Journal of Zoology,31(3):358-363.] doi:10.3969/j.issn.1000-7083. 2012.03.005.
李超,侯吉伦,王桂兴,张晓彦,刘永富,童爱萍,刘海金. 2015. 基于牙鲆RNA-seq数据中SSR标记的信息分析[J]. 海洋渔业,37(2):122-127. [Li C,Hou J L,Wang G X,Zhang X Y,Liu Y F,Tong A P,Liu H J. 2015. Bioinformatic analysis of SSR markers in transcriptomic sequenceing Paralichthys olivaceus[J]. Marine Fisheries,37(2):122-127.] doi:10.13233/j.cnki.mar.fish.2015. 02.004.
李清莹,仲崇禄,姜清彬,张勇,陈羽,魏永成,陈珍. 2019. 珍贵树种火力楠转录组SSR特征分析[J]. 基因组学与应用生物学,38(4):1674-1682. [Li Q Y,Zhong C L,Jiang Q B,Zhang Y,Chen Y,Wei Y C,Chen Z. 2019. Characteri-stic analysis of microsatellites in the transcriptome of Michelia macclurei of rare tree species[J]. Genomics and Applied Biology,38(4):1674-1682.] doi:10.13417/j.gab. 038.001674.
梁霞,王慧琪,马宇璇,宋磊,吴超,李亮徽,张国松. 2021. 鲤鱼(Cyprinus carpio)全基因组微卫星分布特征研究[J]. 南京师大学报(自然科学版),44(3):103-111. [Liang X,Wang H Q,Ma Y X,Song L,Wu C,Li L H,Zhang G S. 2021. Distribution characteristics of microsatellites in the whole genome of Cyprinus carpio,Linnaeus[J]. Journal of Nanjing Normal University (Natural Science Edition),44(3):103-111.] doi:10.3969/j.issn.1001-4616.2021.03. 016.8C9BE7E2-0628-425C-8B6F-560A759F3A42
刘洪涛,刘金叶,杨明秋,何玉贵,王永波. 2020. 基于转录组测序的波纹唇鱼SSR和SNP多态特征分析[J]. 基因组学与应用生物学,39(6):2451-2461. [Liu H T,Liu J Y,Yang M Q,He Y G,Wang Y B. 2020. SSR and SNP polymorphic feature analysis based on Cheilinus undulatus transcriptome[J]. Genomics and Applied Biology,39(6):2451-2461.] doi:10.13417/j.gab.039.002451.
刘磊,彭士明,高权新,张晨捷,施兆鸿. 2016. 基于银鲳RNA-seq数据中SSR标记的信息分析[J]. 安徽农业科学,44(28):102-105. [Liu L,Peng S M,Gao Q X,Zhang C J,Shi Z H. 2016. Bioinformatic analysis of SSR markers based on RNA-seq of Pampus argenteus[J]. Journal of Anhui Agricultural Sciences,44(28):102-105.] doi:10. 13989/j.cnki.0517-6611. 2016.28.033.
马秋月,戴晓港,陈赢男,张得芳,廖卓毅,李淑娴. 2013. 枣基因组的微卫星特征[J]. 林业科学,49(12):81-87. [Ma Q Y,Dai X G,Chen Y N,Zhang D F,Liao Z Y,Li S X. 2013. Characterization of microsatellites in the genome of Ziziphus jujuba[J]. Scientia Silvae Sinicae,49(12):81-87.] doi:10.11707 /j.1001-7488.20131212.
倪守胜,杨钰,柳淑芳,庄志猛. 2018. 基于高通量测序的虾夷扇贝基因组微卫星特征分析[J]. 渔业科学进展,39(1):107-113. [Ni S S,Yang Y,Liu S F,Zhuang Z M. 2018. Microsatellite analysis of Patinopecten yessoensis using next-generation sequencing method[J]. Progress in Fi-shery Sciences,39(1):107-113.] doi:10.11758/yykxjz. 20161209001.
孙海林,孙成飞,董浚键,田园园,胡婕,叶星. 2019. 翘嘴鳜转录组测序及SSR新标记的开发与应用[J]. 基因组学与应用生物学,38(10):4413-4421. [Sun H L,Sun C F,Dong J J,Tian Y Y,Hu J,Ye X. 2019. Transcriptome sequen-cing and development and application of novel SSR markers for Siniperca chuatsi[J]. Genomics and Applied Biology,38(10):4413-4421.] doi:10.13417/j.gab.038. 004413.
孫赛红. 2014. 红鳍东方鲀4个免疫基因的原核表达及群体EST-SSRs分析[D]. 大连:大连海洋大学. [Sun S H. 2014. Prokaryotic expression analysis of four immune genes and analysis of group EST-SSRs in Takifugu rubri pes[D]. Dalian:Dalian Ocean University.] doi:10.7666/d.D495822.
熊良伟,王帅兵,岳丽佳,王建国,陶桂庆,徐亮,王权. 2018. 宽体金线蛭基因组SSR序列特征分析及其分子标记开发[J]. 南方农业学报,49(11):2298-2303. [Xiong L W,Wang S B,Yue L J,Wang J G,Tao G Q,Xu L,Wang Q. 2018. SSR sequence characters for genome of Whitmania pigra Whitman and development of molecular markers[J]. Journal of Southern Agriculture,49(11):2298-2303.] doi:10.3969/j.issn.2095-1191.2018.11.26.
徐杰杰,毕宜慧,程景颢,邢秀梅,暨杰,王涛,尹绍武,张凯. 2021. 中华绒螯蟹(Eriocheir sinensis)全基因组微卫星分布特征研究[J]. 基因组学与应用生物,40(7-8):2422-2429. [Xu J J,Bi Y H,Cheng J H,Xing X M,Ji J,Wang T,Yin S W,Zhang K. 2021. Study on distribution characteristics of whole genome microsatellite of Eriocheir sinensis[J]. Genomics and Applied Biology,40(7-8):2422-2429.] doi:10.13417/j.gab.040.002422.
杨芩,付燕,刘雅兰,张婷渟,彭舒,邓洁. 2021. 蓝莓花粉转录组SSR位点信息分析[J]. 分子植物育种,19(10):3383-3391. [Yang Q,Fu Y,Liu Y L,Zhang T T,Peng S,Deng J. 2021. Analysis of SSR information in Bluebery pollen transcriptome[J]. Molecular Plant Breeding,19(10):3383-3391.]. doi:10.13271/j.mpb.019.003383.8C9BE7E2-0628-425C-8B6F-560A759F3A42
岳華梅,翟晴,宋明月,叶欢,杨晓鸽,李创举. 2016. 基于转录组测序的兴国红鲤微卫星标记筛选[J]. 淡水渔业,46(1):24-28. [Yue H M,Zhai Q,Song M Y,Ye H,Yang X G,Li C J. 2016. Development of microsatellite mar-kers in Cyprinus carpio var. singuonensis using next-ge-neration sequencing[J]. Freshwater Fisheries,46(1):24-28.] doi:10.13721/j.cnki.dsyy.2016.01.004.
赵燕,陈红菊,孔维祎,季相山,王慧. 2020. 日本沼虾多态性标记筛选及群体遗传结构分析[J]. 水产科学,39(5):639-648. [Zhao Y,Chen H J,Kong W Y,Ji X S,Wang H. 2020. Screening of polymorphic markers and analysis of population genetic structure of oriental river prawn Macrobrachium nipponense[J]. Fisheries Science,39(5):639-648.] doi:10.16378/j.cnki.1003-1111.2020.05.001.
Bai Z Y,Yin Y X,Hu S N,Wang G L,Zhang X W,Li J L. 2009. Identification of genes involved in immune response,microsatellite,and SNP markers from expressed sequence tags generated from hemocytes of freshwater pearl mussel(Hyriopsis cumingii)[J]. Marune Biotechnology,11(4):520. doi:10.1007/s10126-008-9163-0.
Borodinsky L N. 2017. Xenopus laevis as a model organism for the study of spinal cord formation,development,function and regeneration[J]. Frontiers in Neural Circuits,11:90. doi:10.3389/fncir.2017.00090.
Chan H K,Shoemaker K T,Karraker N E. 2014. Demography of Quasipaa frogs in China reveals high vulnerability to widespread harvest pressure[J]. Biological Conservation,170:3-9. doi:10.1016/j.biocon.2013.12.014.
Che R B,Sun Y N,Wang R X,Xu T J. 2014. Transcriptomic analysis of endangered Chinese salamander:Identification of immune,sex and reproduction-related genes and genetic markers[J]. PLoS One,9(1):e87940. doi:10.1371/journal.pone.0087940.
Gao Z X,Luo W,Liu H,Zeng C,Liu X L,Yi S K,Wang W M. 2012. Transcriptome analysis and SSR/SNP markers information of the blunt snout bream(Megalobrama amblycephala)[J]. PLoS One,7(8):e42637. doi:10.1371/journal.pone.0042637.
Garrido-Cardenas J A,Mesa-Valle C,Manzano-Agugliaro F. 2018. Trends in plant research using molecular marker[J]. Planta,247(3):543-557. doi:10.1007/s00425-017-2829-y.
Harr B,Schl?tterer C. 2000. Long microsatellite alleles in Drosophila melanogaster have a downward mutation bias and short persistence times,which cause their genome-wide underrepresentation[J]. Genetics,155(3):1213-1220. doi:10.1093/genetics/155.3.1213.
Hellsten U,Harland R M,Gilchrist M J,Hendrix D,Jurka J,Kapitonov V,Ovcharenko I,Putnam N H,Shu S,Taher L,Blitz I L,Blumberg B,Dichmann D S,Dubchak I,Amaya E,Detter J C,Fletcher R,Gerhard D S,Goodstein D,Graves T,Grigoriev I V,Grimwood J,Kawashima T,Lindquist E,Lucas S M,Mead P E,Mitros T,Ogino H,Ohta Y,Poliakov A V,Pollet N,Robert J,Salamov A,Sater A K,Schmutz J,Terry A,Vize P D,Warren W C,Wells D,Wills A,Wilson R K,Zimmerman L B,Zorn A M,Grainger R,Grammer T,Khokha M K,Richardson P M,Rokhsar D S. 2010. The genome of the Western clawed frog Xenopus tropicalis[J]. Science,328(5978):633-636. doi:10.1126/science.1183670.8C9BE7E2-0628-425C-8B6F-560A759F3A42
Hsu T H,Chiu Y T,Lee H T,Gong H Y,Huang C W. 2021. Development of EST-molecular markers from RNA sequencing for genetic management and identification of growth traits in potato grouper(Epinephelus tukula)[J]. Biology (Basel),10(1):36. doi:10.3390/biology10010 036.
Hu W F,Dong B J,Kong S S,Mao Y Y,Zheng R Q. 2017. Pathogen resistance and gene frequency stability of major histocompatibility complex class IIB alleles in the giant spiny frog Quasipaa spinosa[J]. Aquaculture,468:410-416. doi:10.1016/j.aquaculture.2016.11.001.
Huang Y,Xiong J L,Gao X C,Sun X H. 2017. Transcriptome analysis of the Chinese giant salamander (Andrias davidianus ) using RNA-sequencing[J]. Genomics Data,14:126-133. doi:10.1016/j.gdata.2017.10.005.
Jo E,Lee S J,Choi E,Kim J,Lee S G,Lee J H,Kim J H,Park H. 2021. Whole genome survey and microsatellite motif identification of Artemia franciscana[J]. Bioscie-nce Reports,41(3):BSR20203868. doi:10.1042/BSR20 203868.
Li B,Dewey C N. 2011. RSEM:Accurate transcript quantification from RNA-Seq data with or without a reference genome[J]. BMC Bioinformatics,12:323. doi:10.1186/1471-2105-12-323.
Li C J,Ling Q F,Ge C,Ye Z Q,Han X F. 2015. Transcriptome characterization and SSR discovery in large-scale loach Paramisgurnus dabryanus(Cobitidae,Cypriniformes)[J]. Gene,557(2):201-208. doi:10.1016/j.gene. 2014.12.034.
Mable B K,Alexandrou M A,Taylor M I. 2011. Genome duplication in amphibians and fish:An extended synthesis[J]. Journal of Zoology,284(3):151-182. doi:10.1111/j. 1469-7998.2011.00829.x.
Pandey M,Kumar R,Srivastava P,Agarwal S,Srivastava S,Nagpure N S,Jena J K,Kushwaha B. 2018. WGSSAT:A high-throughput computational pipeline for mining and annotation of SSR markers from whole genomes[J]. Journal of Heredity,109(3):339-343. doi:10.1093/jhered/esx075.
Rhode C,Roodt-Wilding R. 2011. Bioinformatic survey of Haliotis midae microsatellites reveals a non-random distribution of repeat motifs[J]. Biological Bulletin,221(2):147-154. doi:10.1086/BBLv221n2p147.
Savage A E,Kiemnec-Tyburczy K M,Ellison A R,Fleischer R C,Zamudio K R. 2014. Conservation and divergence in the frog immunome:Pyrosequencing and de novo assembly of immune tissue transcriptomes[J]. Gene,542(2):98-108. doi:10.1016/j.gene.2014.03.051.
Trick M,Long Y,Meng J L,Bancroft I. 2009. Single nucleotide polymorphism (SNP) discovery in the polyploidy Brassica napus using solexa transcriptome sequencing[J]. Plant Biotechnol Journal,7(4):334-346. doi:10.1111/j.1467-7652. 2008.00396.x.8C9BE7E2-0628-425C-8B6F-560A759F3A42
Tsai H Y,Hamilton A,Guy D R,Tinch A E,Bishop S C,Houston R D. 2015.Verification of SNPs associated with growth traits in two populations of farmed Atlantic salmon[J]. International Journal of Molecular Sciences,17(1):5. doi:10.3390/ijms17010005.
Wang D,Liao X L,Cheng L,Yu X M,Tong J G. 2007. Deve-lopment of novel EST-SSR markers in common carp by data mining from public EST sequences[J]. Aquaculture,271(1-4):558-574. doi:10.1016/j.aquaculture.2007.06. 001.
Wang Y,Yang L D,Wu B,Song Z B,He S P. 2015. Transcriptome analysis of the plateau fish (Triplophysa dalaica):Implications for adaptation to hypoxia in fishes[J]. Gene,565(2):211-220. doi:10.1016/j.gene.2015.04.023.
Xia Y,Luo W,Yuan S Q,Zheng Y C,Zeng X M. 2018. Microsatellite development from genome skimming and transcriptome sequencing:Comparison of strategies and lessons from frog species[J]. BMC Genomics,19(1):886. doi:10.1186/s12864-018-5329-y.
Yá?ez J M,Yoshida G,Barria A,Palma-Véjares R,Travisany D,Díaz D,Cáceres G,Cádiz M I,López M E,Lhorente J P,Jedlicki A,Soto J,Salas D,Maass A. 2020. High-throughput single nucleotide polymorphism(SNP) disco-very and validation through whole-genome resequencing in Nile tilapia(Oreochromis niloticus)[J]. Marine Biotechnology(NY),22(1):109-117. doi:10.1007/s10126-019-09935-5.
Ye S P,Huang H,Zheng R Q,Zhang J Y,Yang G,Xu S X. 2013. Phylogeographic analyses strongly suggest cryptic speciation in the giant spiny frog(Dicroglossidae:Paa spinosa) and interspecies hybridization in Paa[J]. PLoS One,8:e70403. doi:10.1371/journal.pone.0070403.
Yu D D,Zheng R Q,Lu Q F,Yang G,Fu Y,Zhang Y. 2016. Genetic diversity and population structure for the conservation of giant spiny frog (Quasipaa spinosa) using microsatellite loci and mitochondrial DNA[J]. Asian Herpetological Research,7(2):75-86. doi:10.16373/j.cnki.ahr. 150040.
Zhang J,Ma W G,Song X M,Lin Q H,Gui J F,Mei J. 2014. Characterization and development of EST-SSR markers derived from transcriptome of yellow catfish[J]. Molecules,19(10):16402-16415. doi:10.3390/molecules1910 16402.
Zhang L L,Bao Z M,Wang S,Hu X L,Hu J J. 2008. FISH mapping and identification of Zhikong scallop(Chlamys farreri) chromosomes[J]. Marine Biotechnology(NY),10(2):151-157. doi:10.1007/s10126-007-9045-x.
Zhang S Y,Li J,Qin Q,Liu W,Bian C,Yi Y H,Wang M H,Zhong L Q,You X X,Tang S K,Liu Y S,Huang Y,Gu R B,Xu J M,Bian W J,Shi Q,Chen X H. 2018. Whole-genome sequencing of Chinese yellow catfish provides a valuable genetic resource for high-throughput identification of toxin genes[J]. Toxins (Basel),10(12):488. doi:10.3390/toxins10120488.
Zhao H,Fuller A,Thongda W,Mohammed H,Abernathy J,Beck B,Peatman E. 2019. SNP panel development for genetic management of wild and domesticated white bass (Morone chrysops)[J]. Animal Genetics,50(1):92-96. doi:10.1111/age.12747.
Zhao X Y,Tan Z Y,Feng H P,Yang R H,Li M F,Jiang J H,Shen G L,Yu R Q. 2011. Microsatellites in different Potyvirus genomes:Survey and analysis[J]. Gene,488(1-2):52-56. doi:10.1016/j.gene.2011.08.016.
Zheng R Q,Ye R H,Yu YY,Yang G. 2009. Fifteen polymorphic microsatellite markers for the giant spiny frog,Paa spinosa[J]. Molecular Ecology Resources,9(1):336-368. doi:10.1111/j.1755-0998.2008.02420.x.
(責任编辑 兰宗宝)8C9BE7E2-0628-425C-8B6F-560A759F3A42
猜你喜欢
江苏农业科学(2016年11期)2017-03-21 07:38:20
科技创新与应用(2016年36期)2017-02-21 20:08:49
安徽农学通报(2017年1期)2017-02-15 18:21:58
中国医药导报(2016年28期)2017-01-06 19:52:54
农业与技术(2016年20期)2016-12-08 21:37:07
赤峰学院学报·哲学社会科学版(2015年11期)2015-12-10 14:13:58
湖北农业科学(2015年18期)2015-10-13 21:27:08
河北渔业(2015年9期)2015-09-15 11:10:08
天津农业科学(2015年7期)2015-09-07 00:00:47
河北渔业(2015年6期)2015-06-24 10:58:32
