
36%氟啶·噻虫啉悬浮剂的高效液相色谱分析方法
2022-06-10 01:40孙雪莹田海霞郭明杰
河北北方学院学报(自然科学版) 2022年5期
孙雪莹,田海霞,郭明杰
(1.上海萤火虫农业科技中心,上海 201799;2.兴农药业(中国)有限公司,上海 201416)
氟啶虫酰胺(flonicamid)是一种高选择性杀虫剂,主要用于防治刺吸式口器害虫,原理是通过抑制昆虫内向整流钾离子(Kir)通道,干扰昆虫细胞的离子稳态与平衡电位,破坏马氏管与唾液腺的正常分泌功能,从而影响昆虫的取食与排泄过程,最终导致害虫死亡[1]。噻虫啉(thiacloprid)是一种新烟碱类杀虫剂,主要用于防治刺吸式口器和部分咀嚼式口器害虫,具有高效、广谱、低毒和高选择性等优点[2],对刺吸式口器害虫、鞘翅目等害虫具有十分突出的防治效果,成为当今使用最为广泛的杀虫剂之一。噻虫啉作为新烟碱类杀虫剂中对蜜蜂较安全的药剂成为吡虫啉、噻虫嗪、噻虫胺等的替代品,近几年在我国大量生产并推广使用[3]。将氟啶虫酰胺和噻虫啉复配,可有效防治蚜虫、粉虱、各种甲虫(如马铃薯甲虫、苹果象甲、稻象甲)和鳞翅目害虫(如苹果树上潜叶蛾和苹果蠹蛾)等,应用前景广阔。而关于氟啶虫酰胺和噻虫啉的检测方法报道主要是气相色谱法和液相色谱法[4-11],本文采用反相高效液相色谱法,在同一色谱条件下对36%氟啶·噻虫啉悬浮剂有效成分进行定量分析。该方法操作简便、快速,准确度和精密度良好,适用于产品质量的检测分析。
1 试验方法
1.1 仪器与试剂
Agilent1260型高效液相色谱仪配有可变波长检测器、自动进样器和Agilent色谱工作站。
水为新蒸二次蒸馏水;甲醇为色谱纯;氟啶虫酰胺标样:纯度≥98.0%;噻虫啉标样:纯度≥98.0%;36%氟啶·噻虫啉悬浮剂(标样和样品均由兴农药业(中国)有限公司提供)。
1.2 色谱条件
色谱柱:采用Zorbax Eclipse XDB-C18(250 mm×4.6 mm i.d.,5μm);检测波长:260 nm;流动相:甲醇∶水=45∶55(体积比);流速:1.0 mL/min;进样量:5 μL;柱温:30 ℃。在上述色谱条件下,噻虫啉和氟啶虫酰胺的保留时间分别约为3.7 min和7.2 min(图1~2)。
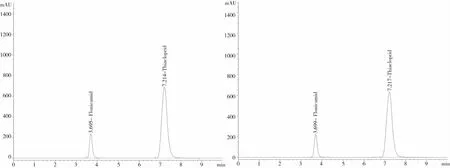
图1 噻虫啉+氟啶虫酰胺标样液相色谱 图2 36%氟啶·噻虫啉悬浮剂试样液相色谱
1.3 溶液配制
1.3.1 标样溶液的配制
称取氟啶虫酰胺标样约0.05 g、噻虫啉标样约0.10 g(精确至0.000 1 g)置于100 mL容量瓶中,用甲醇溶解,于超声波中振荡至完全溶解,取出,恢复至室温后用甲醇定容至刻线摇匀,备用。
1.3.2 试样溶液的配制
称取36%氟啶·噻虫啉悬浮剂试样0.41 g(精确至0.000 1 g),置于100 mL容量瓶中,用甲醇溶解,于超声波中振荡至完全溶解,取出,恢复至室温后用甲醇定容至刻线摇匀,用0.22 μm有机滤膜过滤后备用。
1.4 测定
在上述液相色谱操作条件下,待仪器基线稳定后,连续注入数针标样溶液,计算各针相对响应值的重复性,待相邻两针的相对响应值变化小于1.0%,按下列顺序进样分析:标样溶液,试样溶液,试样溶液,标样溶液。用外标法计算各自含量。
1.5 计算
将测得的2针试样溶液以及试样前后2针标样溶液中的氟啶虫酰胺(或噻虫啉)峰面积分别进行平均。试样中氟啶虫酰胺(或噻虫啉)的质量分数ω(%)按照公式(1)计算:
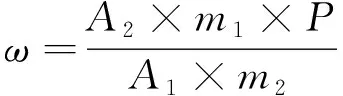
(1)
式中:A1为标样溶液中氟啶虫酰胺(或噻虫啉)峰面积的平均值;A2为试样溶液中氟啶虫酰胺(或噻虫啉)峰面积的平均值;m1为标样质量(g);m2为试样质量(g);P为标样中氟啶虫酰胺(或噻虫啉)的质量分数,%(m/m)。
2 实验过程及步骤
2.1 色谱条件的选择
2.1.1 流动相的选择
流动相的选择是决定液相色谱分离效果的关键所在。通过试用不同比例的甲醇、水,甲醇、水作为流动相对试样进行分离检测。根据检测结果,采用甲醇、水作为流动相分离效果好,出峰时间短,且峰型尖锐,最终选定流动相的比例为甲醇∶水=45∶55(体积比)。
2.1.2 检测波长的选择
通过对氟啶虫酰胺和噻虫啉进行紫外扫描,得到其相应的吸收波长与响应值的紫外吸收光谱图。当选用260 nm作为检测波长时,氟啶虫酰胺和噻虫啉均有较强吸收峰,同时杂质响应值小,流动相无吸收。因此,为兼顾两者均有较好的吸收,将检测波长选为260 nm。
2.2 特异性的测定
本试验方法采用HPLC-DAD峰纯度分析法来鉴别。氟啶虫酰胺HPLC-DAD中的最小峰纯度相似度均为0.999 846,最小峰纯度阈值为0.999 964,最小峰纯度指数为正值,说明样品色谱峰中不含有杂质峰;噻虫啉HPLC-DAD中的最小峰纯度相似度为0.999 999,最小峰纯度阈值为0.999 999,最小峰纯度指数为正值,说明样品色谱峰中不含有杂质峰。标样液相色谱峰与试样液相色谱峰出峰时间差在1.0%以内,如图1~2所示。制剂中峰纯度色谱图如图3~4所示。
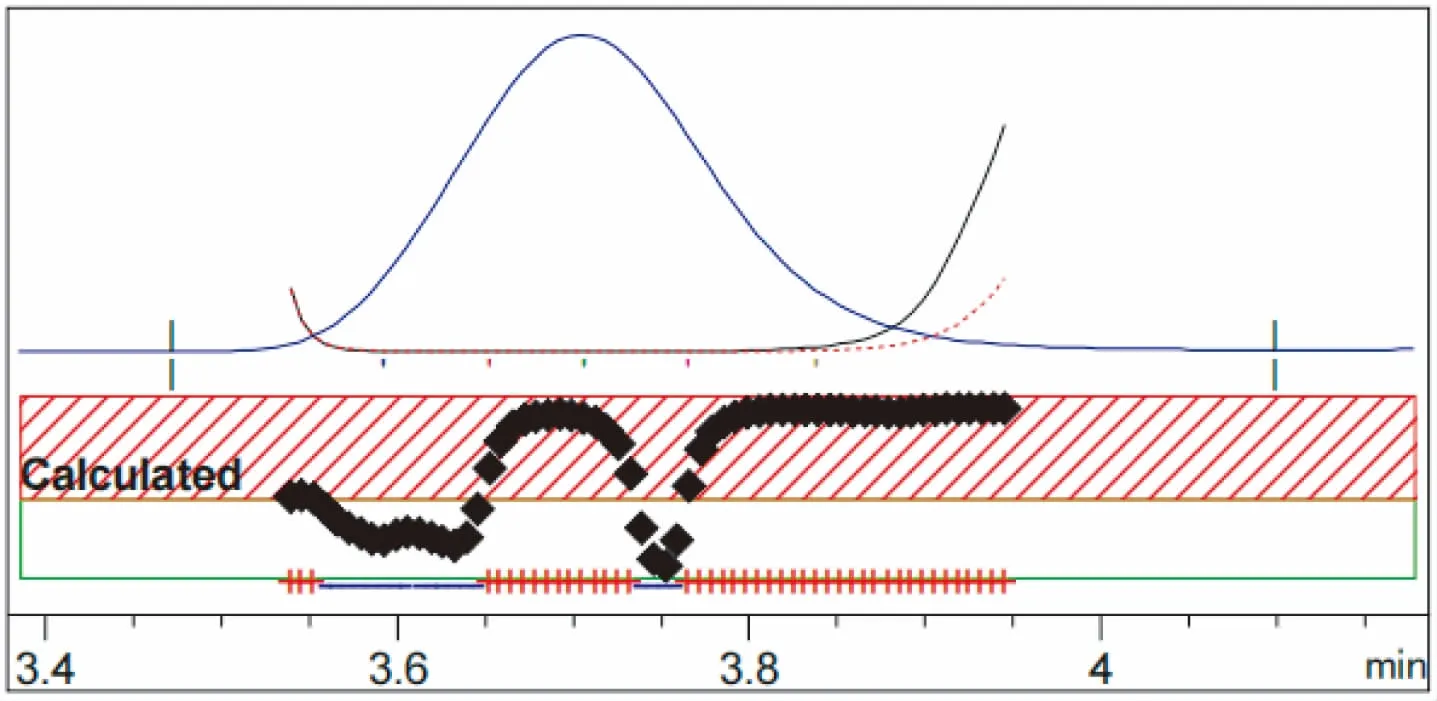
图3 制剂中氟啶虫酰胺HPLC-DAD峰纯度色谱
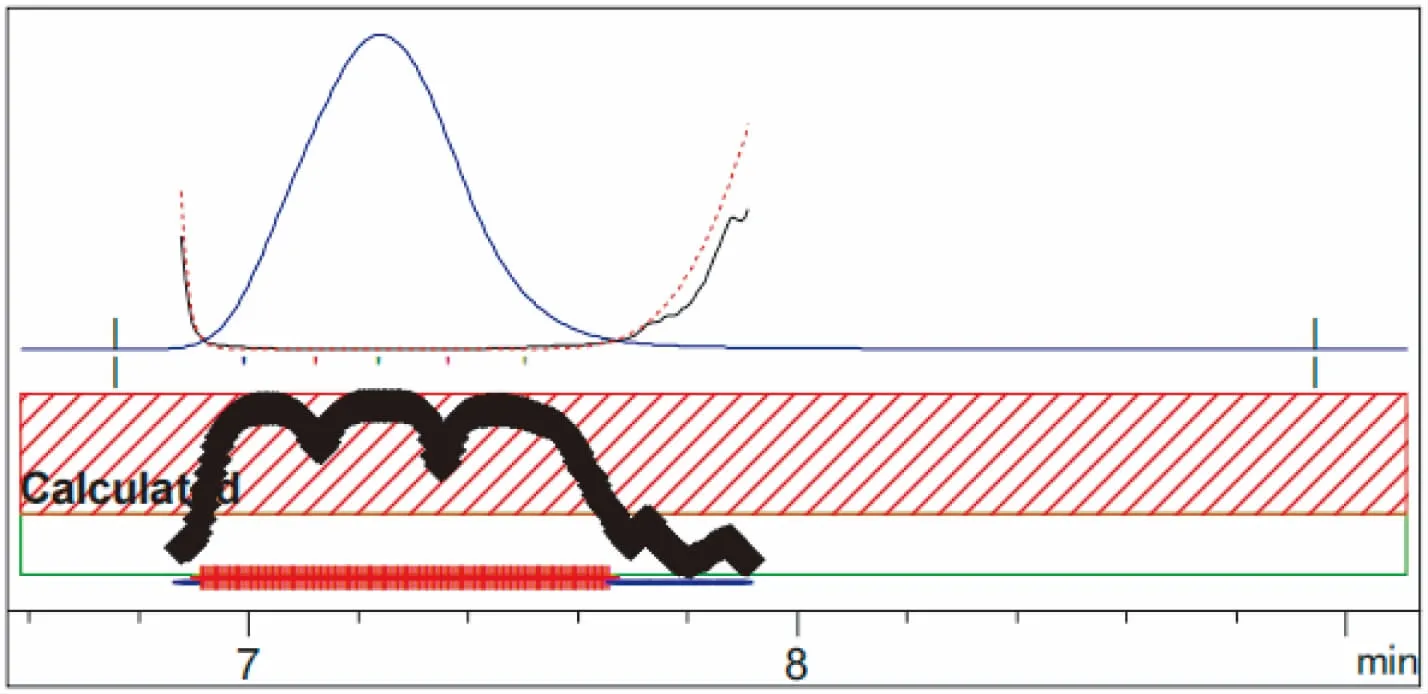
图4 制剂中噻虫啉HPLC-DAD峰纯度色谱
2.3 线性相关性测定
在一定质量范围内,按照浓度递增的顺序配制数个标样,按标准规定的操作步骤进行分析,测定氟啶虫酰胺/噻虫啉的峰面积,取两次测定的平均结果。以氟啶虫酰胺/噻虫啉标样溶液之浓度(mg·L-1)为横坐标,峰面积(mAU)为纵坐标绘制标准曲线,从而得到氟啶虫酰胺和噻虫啉的线性相关性曲线。氟啶虫酰胺线性方程是:y=4.6863x-24.303(图5),相关系数为1.000 0;噻虫啉的线性方程是:y=13.031x-8.51(图6),相关系数为1.000 0。
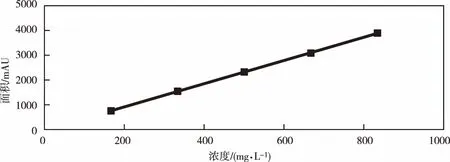
图5 氟啶虫酰胺线性关系
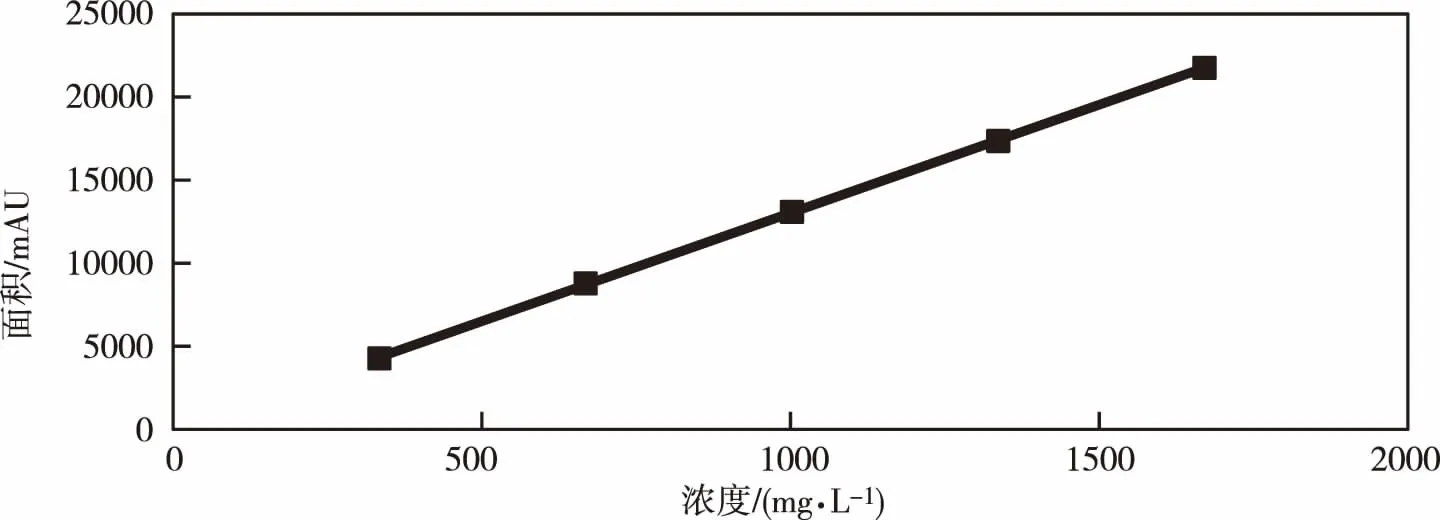
图6 噻虫啉线性关系
2.4 分析方法的精密度测定
在上述色谱操作条件下,对同一36%氟啶·噻虫啉悬浮剂样品平行测定5次,氟啶虫酰胺和噻虫啉的标准偏差分别为0.001 4和0.002 2,变异系数分别为1.16%和1.66%,结果见表1。
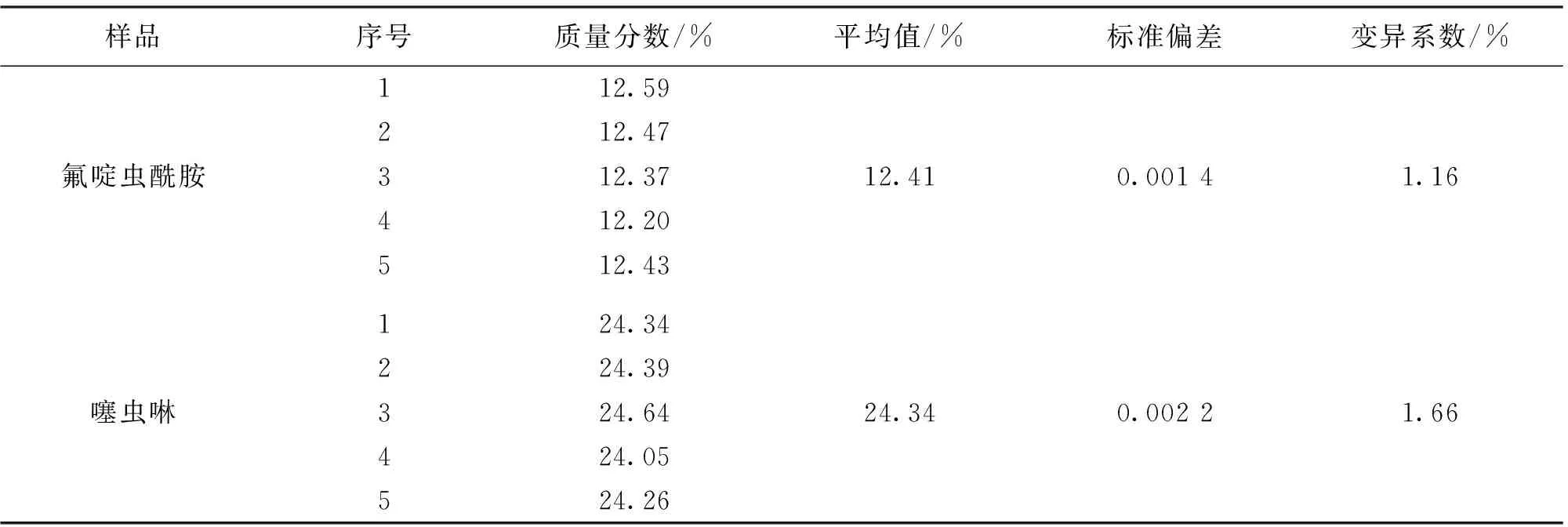
表1 36%氟啶·噻虫啉悬浮剂分析方法的精密度试验结果
2.5 分析方法的准确度测定
制剂空白:按配方比例要求,除氟啶虫酰胺原药、噻虫啉原药外将所有助剂混合均匀。在制剂空白样品中分别加入不同质量已知含量的氟啶虫酰胺标样、噻虫啉标样至4个于100 mL容量瓶中,加入60 mL甲醇,置于超声波中超声振荡至完全溶解,取出,恢复至室温后用甲醇定容至刻线,摇匀,用0.22 μm有机滤膜过滤后备用。
按照上述色谱条件测定样品的氟啶虫酰胺和噻虫啉总浓度,并计算其平均回收率,结果见表2。氟啶虫酰胺和噻虫啉的平均回收率分别为99.88%和99.91%。

表2 36%氟啶·噻虫啉悬浮剂分析方法的回收率试验结果
2.6 非分析物干扰的试验
在评价准确度时,通常需要包含非分析物质的干扰,因为有效成分中的任何干扰物都会导致分析方法出现系统误差。而且,在分析时应同时测定不带有效成分的空白样品,或证明其无干扰。
通过对空白样品(不含有效成分的助剂)、原药等按分析方法1.2进行分析,如图7~9所示,各助剂与原药无相互干扰,对分析无干扰,均不影响相互定性定量分析,因此本文的分析方法未受到非分析物干扰。
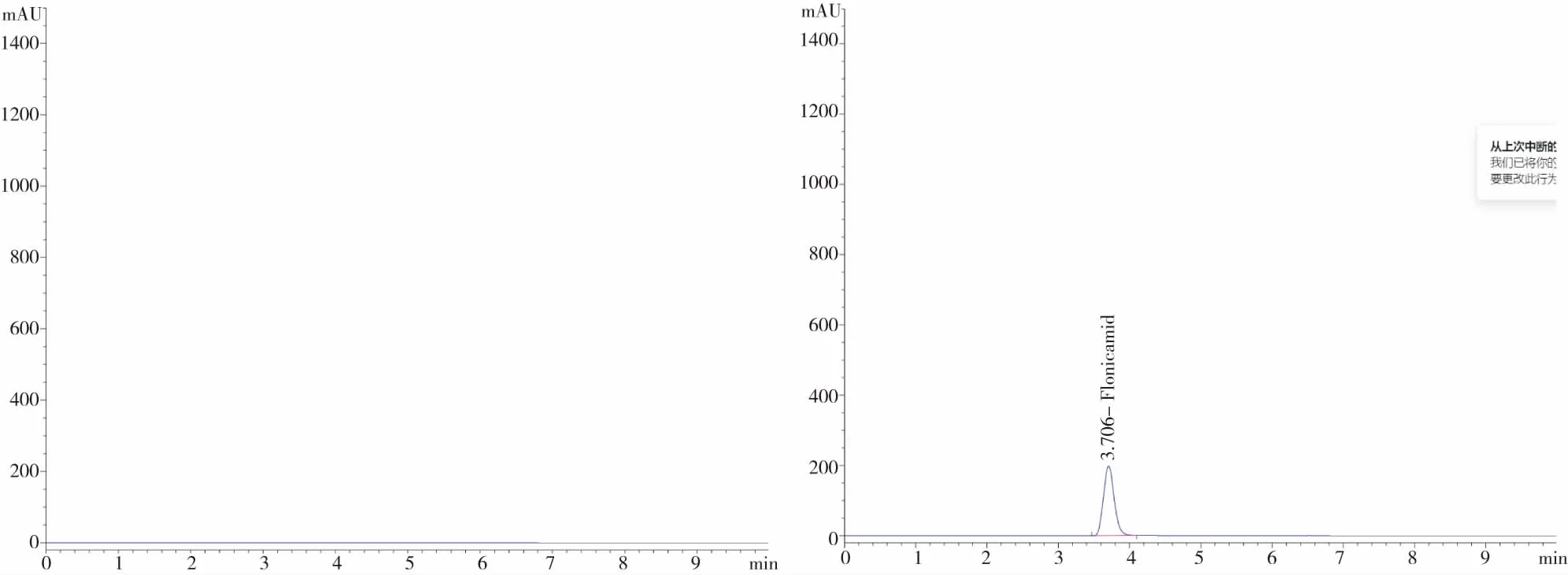
图7 空白样品液相色谱 图8 氟啶虫酰胺原药液相色谱
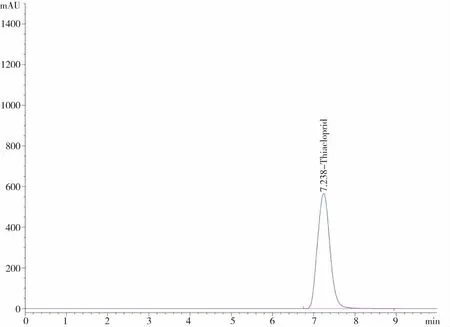
图9 噻虫啉原药液相色谱
3 结 论
本文建立了36%氟啶·噻虫啉悬浮剂中有效成分的分析方法,此方法具有高精密度和高准确度的特点,其线性关系良好,试验操作简便,是进行在线产品质量检测较理想的分析方法。
猜你喜欢
理化检验-化学分册(2022年11期)2022-11-27
分子催化(2022年1期)2022-11-02
农药科学与管理(2022年6期)2022-08-03
粉末冶金技术(2021年3期)2021-07-28
磷肥与复肥(2021年5期)2021-06-19
——第二部分:原棉短纤维率标样的验证试验分析
中国纤检(2020年7期)2020-07-22
当代水产(2020年3期)2020-06-15
农药科学与管理(2019年12期)2019-05-20
食品界(2018年8期)2018-09-03
中学生数理化·高二版(2016年6期)2016-05-14
