
HPLC-CAD同时测定复方醋酸钠林格注射液中钠、钾、镁、钙的含量
2022-06-10 12:16钱广生
中国测试 2022年5期
杨 琴,袁 军,刘 峰,钱广生
(1. 四川大学华西药学院,四川 成都 610041; 2. 四川省药品检验研究院,四川 成都 611730)
0 引 言
复方醋酸钠林格注射液中含有多种电解质(Na+、K+、Mg2+、Ca2+、Cl–、Acetate–、Gluconate–、Citrate3–)和葡萄糖,在临床上用于循环血量及组织间液减少时的细胞外液的补充,代谢性酸中毒的纠正[1],也应用于各类型手术中补液[2-3]。为了临床上电解质输入的准确性和科学性,需要对复方醋酸钠林格注射液中的各电解质含量进行测定。目前其质量标准中测定各离子的方法为原子吸收光谱法,需对其中的每一种离子进行单独分析,耗时长、成本高。除原子吸收光谱法外,离子色谱-电导检测器也被用于样品中钠、钾、镁、钙的测定[4],但电导检测器的响应受温度影响较大。在其他领域,样品中的钠、钾、镁、钙常采用电感耦合等离子体质谱法(ICPMS)[5],但ICP-MS较昂贵,对氩气消耗大,耗材更换频率高。电雾式检测器(charged aerosol detector ,CAD)作为一种新型的通用型质量型检测器[6],现已被2020年版《中国药典》收载。CAD的响应值只与进样质量有关,不依赖于化合物的结构与性质,具有检测范围宽、灵敏度高、重现性好的优势[7-8],可应用于中性、酸性、碱性及两性物质等,特别是无紫外吸收、非挥发性或半挥发性物质的检测[9]。有文献报道,利用CAD检测器同时测定葡萄糖氯化钠注射液中葡萄糖、氯和钠的含量[10]。为了缩短测定时间,本实验采用阳离子分析柱与通用型检测器——CAD,同时测定其中钠、钾、镁、钙离子的含量,能更加快速、简便、准确地对复方醋酸钠林格注射液进行定量分析。
1 实验部分
1.1 仪器与试药
Vanquish高效液相色谱仪(美国Thermo);电雾式检测器(美国Thermo);Arium pro超纯水仪(德国Sartorius);复方醋酸钠林格注射液,批号为D20122309(方法学验证批)、T20082901、T20083001,四川科伦药业股份有限公司;钠元素标准溶液(批号208043-3);钾元素标准溶液(批号208042-2);镁元素标准溶液(批号194013-3);钙元素标准溶液(批号201007-4);乙腈(新蓝景,色谱纯);三氟乙酸(安谱,色谱纯)。
1.2 方法与结果
1.2.1 HPLC-CAD条件
采用SiELC Primesep A(4.6 mm×250 mm,5 μm)色谱柱,柱温30 ℃,流动相A为0.1%三氟乙酸水溶液,流动相B为0.25%三氟乙酸乙腈溶液,按表1中的程序进行梯度洗脱,流量1 mL/min,进样器温度15 ℃,进样量25 μL。采用CAD检测器,数据采集频率10 Hz,滤波5 s,雾化器温度50 ℃,幂函数1。

表1 流动相洗脱程序
1.2.2 溶液的制备
精密量取样品2 mL于10 mL容量瓶中,加水稀释至刻度,摇匀,作为供试品溶液A,供测定钾、钙、镁离子。精密量取1 mL样品于100 mL容量瓶中,加水稀释至刻度,摇匀,作为供试品溶液B,供测定钠离子。分别精密量取钠、钾、镁、钙单元素标准溶液(1 000 μg/mL)1 mL 于 10 mL 容量瓶中,加水稀释至刻度,摇匀,作为钠、钾、镁、钙单元素对照品贮备液。分别精密量取钠、钾、镁、钙单元素对照品贮备液3 mL,3 mL,0.5 mL,1.2 mL于同一10 mL容量瓶中,加水稀释至刻度,摇匀,制成浓度分别为30 μg/mL,30 μg/mL,5 μg/mL,12 μg/mL 的对照品溶液。
1.2.3 专属性
精密量取空白溶剂、对照品溶液、供试品溶液25 μL,分别进样测定,记录色谱图,见图1。结果表明:空白溶剂不干扰待测组份测定,钠、钾、镁、钙的保留时间分别为 7.046 min、9.256 min、17.190 min、19.077 min左右,峰形良好,供试品溶液中无其他杂质峰干扰。

图1 溶液色谱图
1.2.4 线性与范围
分别精密量取钠、钾、镁、钙单元素标准溶液(1 000 μg/mL)3 mL、3 mL、0.5 mL、1.2 mL 于同一25 mL容量瓶中,加水稀释至刻度,摇匀,作为线性储备液。精密量取 1.25,2.5,2.5,2.5,3.75,5 mL 线性储备液,分别置于 50,50,20,10,10,10 mL 容量瓶中,加水稀释至刻度,摇匀,依次等体积按“1.2.1”项下方法进行测定,记录色谱图。以质量浓度的对数为横坐标,峰面积的对数为纵坐标,绘制标准曲线。钠、钾、镁、钙的回归方程与线性范围见表2。
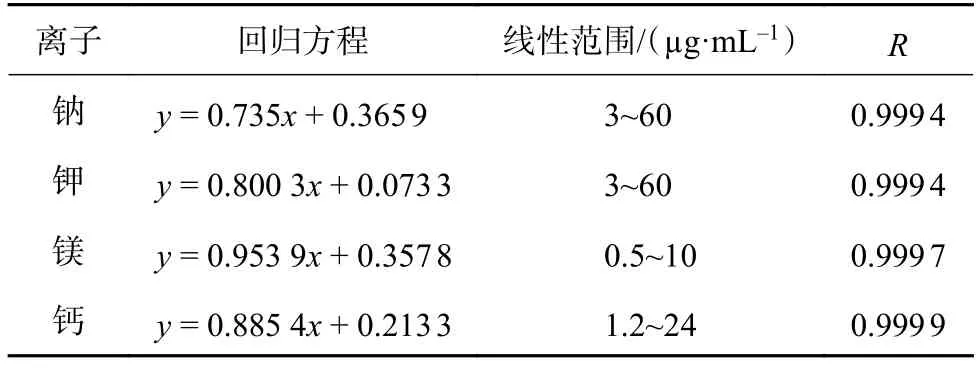
表2 钠、钾、镁、钙的回归方程、线性范围和相关系数
1.2.5 检测限与定量限
精密量取各对照品储备液适量,逐级稀释后进样测定,记录色谱图,以信噪比为3时的质量浓度作为检测限,以信噪比为10时的质量浓度作为定量限。测得钠的检测限为0.154 μg/mL,定量限为0.330 μg/mL;钾的检测限为 0.286 μg/mL,定量限为 0.571 μg/mL;镁的检测限为 0.100 μg/mL,定量限为 0.200 μg/mL;钙的检测限为 0.100 μg/mL,定量限为 0.200μg/mL。
1.2.6 进样精密度与重复性试验
取对照品溶液,连续进样6针,记录峰面积,计算得钠、钾、镁、钙峰面积的RSD分别为0.45%、0.79%、0.69%、0.52%(n=6),说明本方法进样精密度良好,精密度结果见表3。按1.2.2项下方法分别平行制备6份供试品溶液A与供试品溶液B,按1.2.1项下方法进行测定,记录峰面积,计算样品中钠、钾、镁、钙的含量。重复性实验结果见表4,本文方法重复性良好。
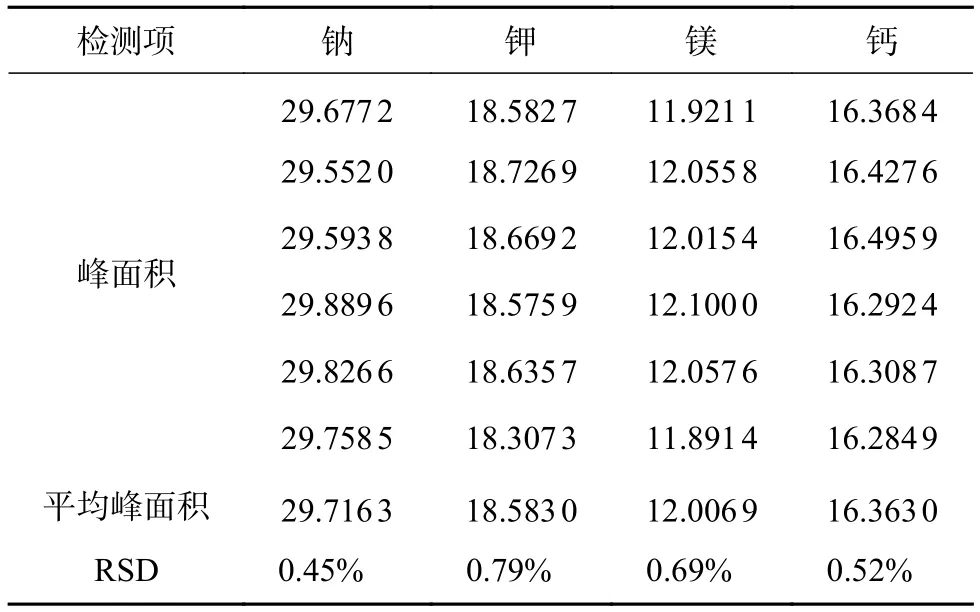
表3 钠、钾、镁、钙的精密度试验结果

表4 钠、钾、镁、钙的重复性试验结果
1.2.7 加样回收试验
精密量取同一样品1 mL,共9份,至10 mL容量瓶中,分别精密加入不同体积的钠对照品储备液,照1.2.2项下方法制备,得80%、100%、120%浓度的加标溶液A。精密量取同一样品0.5 mL,共9份,至100 mL容量瓶中,分别精密加入不同体积的钾、镁、钙对照品储备液,照1.2.2项下方法制备,得80%、100%、120%浓度的加标溶液B。以重复性测定结果为样品本底值,按1.2.1项下方法测定,记录峰面积。计算得钠的回收率在101.0%~102.8%之间,平均回收率为101.7%,RSD为0.70%;钾的回收率在97.19%~100.3%之间,平均回收率为98.41%,RSD为1.05%;镁的回收率在97.38%~101.9%之间,平均回收率为99.28%,RSD为1.48%;钙的回收率在 97.14%~102.2%之间,平均回收率为 99.73%,RSD为1.88%。加样回收试验结果见表5。

表5 钠、钾、镁、钙加样回收率试验
1.2.8 样品测定
取复方醋酸钠林格注射液3批,按1.2.1项下方法进行测定,计算样品中钠、钾、镁、钙的含量,并与标准推荐的原子吸收光谱法进行对比,试验结果见表6,两种方法测定结果相当。
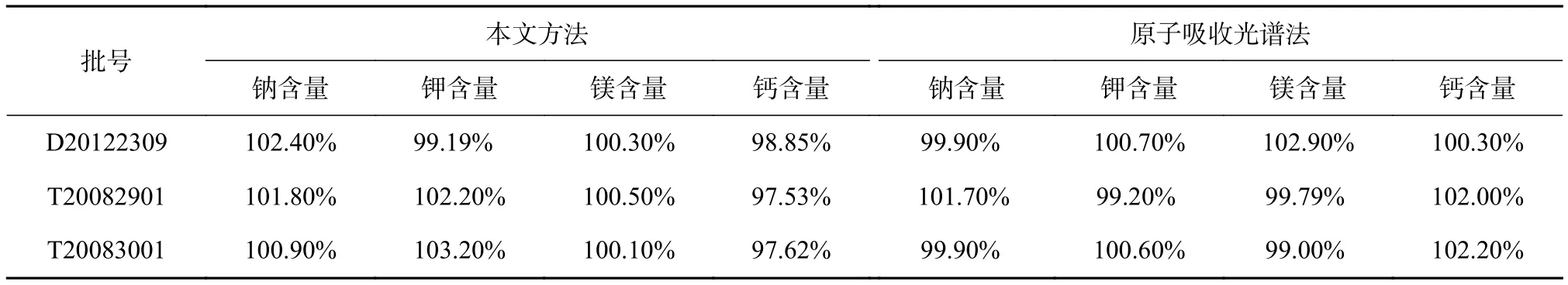
表6 样品中钠、钾、镁、钙的测定结果
1.2.9 耐用性试验
取复方醋酸钠林格注射液(批号D20122309)以及对照品溶液,按表7中更改后的条件进行测定,记录峰面积,计算样品中钠、钾、镁、钙的含量。开发方法时已试验过多根色谱柱,故不再更换色谱柱进行考察。试验结果见表8。7种方法测得的供试品中钠、的钾、镁、钙含量RSD值分别为2.40%、1.60%、0.86%、1.84%,表明方法耐用性良好。

表7 耐用性试验条件
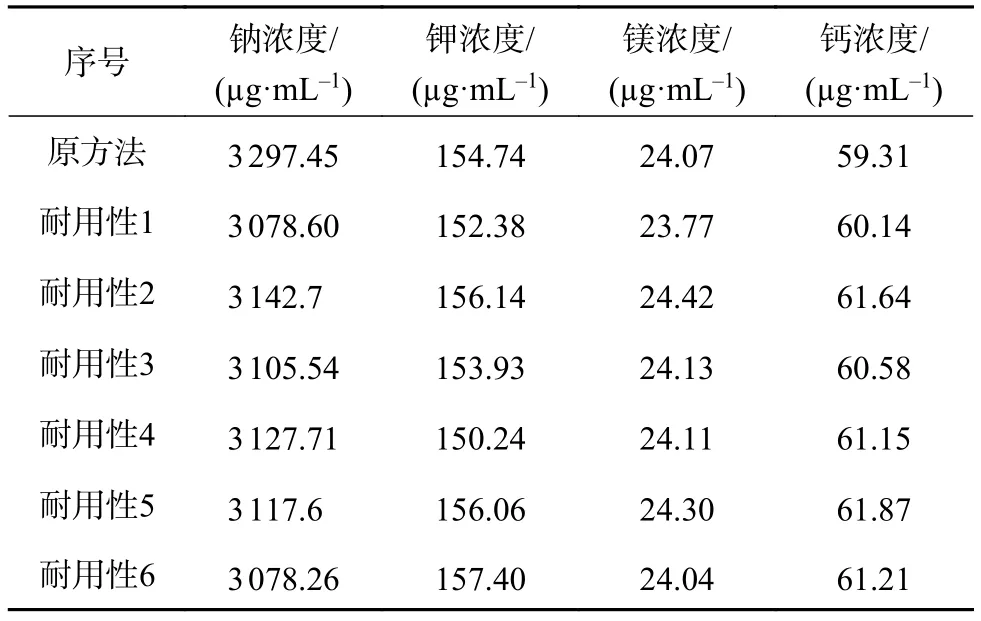
表8 耐用性试验结果
2 讨 论
2.1 供试品溶液浓度的选择
由于样品中钠离子与其他3种离子浓度相差甚大,若用同一份供试品测定4个离子,很难满足钠离子不出现平头峰且其他3个离子达到定量限且峰型良好,所以设计两份稀释倍数不同的供试品溶液A和供试品溶液B;钠离子的供试品溶液A由样品稀释100倍而得,钾、镁和钙离子的供试品溶液B由样品稀释5倍而得,由此得到的色谱图峰形良好。
2.2 色谱条件的优化
本实验考察了多根色谱柱,包括SiELC PrimesepA(4.6 mm×250 mm,5 μm)、Agilent HILIC-Z(3.0 mm×150 mm,2.7 μm)、Thermo Dionex IonPacTMCS12A(4 mm×250 mm),通过比较其对4种阳离子的保留行为和分离度,最终发现SiELC Primesep A (4.6 mm×250 mm,5 μm)柱分离效果最佳。葡萄糖在此色谱条件下率先出峰,但峰形较差,故不对葡萄糖进行分析。
流动相中加入的酸性调节剂选用了具有挥发性的三氟乙酸,并考察了不同浓度三氟乙酸对结果的影响。结果表明,当酸浓度过低时,镁、钙离子不出峰,当浓度过高时,各离子间分离较差,最后发现0.1%三氟乙酸水溶液–0.25%三氟乙酸乙腈系统分离效果最佳,且基线平稳。进一步优化洗脱梯度后,确定了上述的洗脱条件,此时分离效果好,峰形好,理论塔板数高。
3 结束语
本文采用阳离子分析柱和通用型检测器-电雾式检测器,建立了同时测定复方醋酸钠林格注射液中钠、钾、镁、钙离子含量的HPLC-CAD检测方法。本研究对色谱柱和流动相进行了筛选和优化,考察供试品溶液浓度,并进行方法学验证。该方法能较好地分离上述四种离子,不受溶剂和辅料的干扰,灵敏度、精密度、重复性、准确度均良好。该方法测定结果与原子吸收光谱法测定结果无明显差异,可准确定量,且可实现多种离子同时测定,耗时少,操作简便,为金属离子的测定提供了一种新的思路。
猜你喜欢
日用电器(2022年3期)2022-04-14
中国土壤与肥料(2021年5期)2021-12-02
中国应急管理科学(2021年4期)2021-04-13
今日农业(2020年22期)2020-12-14
商品与质量(2020年18期)2020-11-27
中学生数理化·高一版(2020年9期)2020-01-02
中学化学(2019年4期)2019-08-06
火力与指挥控制(2018年10期)2018-11-13
考试周刊(2018年68期)2018-09-17
食品界(2017年7期)2017-08-24
