
锂硫电池正极材料 Fe3O4以及掺杂改性的第一性原理研究
2022-06-09 14:13:52延杜康王自立
黑龙江科学 2022年10期
延杜康,王自立,邓 超
(哈尔滨师范大学 化学化工学院,哈尔滨 150025)
锂硫电池表现出远超出传统锂离子电池的优异性能,更加重要的是电极材料本身无毒和来源广阔的特点可以降低电池的生产成本,因此锂电池装置成了市面上主流移动电源供应产品。但硫阴极在充放电过程中发生的副反应极易导致电池体积膨胀[1]。这些碳材料对于多硫化锂物质的吸附较弱,在将碳材料改进为过渡金属氧化物、过渡金属硫化物和过渡金属磷化物之后表现出对于多硫化锂物质强烈的吸附作用。目前,具有高导电性能和孔隙较大的碳材料已经被广泛应用于硫电极,如常见的空心纳米管、空心纳米方块及多层核壳结构的微米球等。Wang等人采用FeS2做电极,可以提高电池的储存性能[2]。Huang 等人提出N掺杂的CoP,可以增强吸附作用[3]。然而,大多数研究人员对于这些电极材料的电化学性能增强缺乏深入了解,特别是在分子水平上,因此必须通过充分了解界面的相互作用,系统阐述电池材料对于多硫化物的吸附情况。通过掺杂N(或者P等)可以实现针对电极材料电化学性能的改善,增加电极材料自身的导电性能和Li+的迁移能力。
本研究通过操纵N掺杂Fe3O4(N/Fe3O4),设计一款新型的双功能催化剂[4]。研究表明,N掺杂Fe3O4之后,削弱Li2S4的S-S键,促进了放电过程中多硫化合物Li2Sx(x=1、2、4和6)的催化转换[5]。揭示了通过N掺杂Fe3O4可以调控电子态向费米能级移动,增强了导电性,调控了电极表面电子动力学[6]。
1 计算方法
计算基于密度泛函理论,计算模拟在CP2K中进行。采用自旋极化的PBE,交换相关泛函和用于价电子的双ζ-高斯(DZVP)基组。将截断能设置为500 Ry,布里渊区积分采用Gamme近似。过渡态和锂扩散路径的计算采用CI-NEB方法。以Fe3O4(111)面和N/ Fe3O4(111)为slab基底,模拟对多硫化物的吸附过程[7]。
Li2Sx和slab的吸附能用Eads表示,计算方法如下所示:Eads=E(Li2Sx+ slab)-E(slab)-E(Li2Sx)。
其中,E(Li2Sx+ slab)代表Li2Sx/slab的总能,E(slab)和E(Li2Sx)分别代表了slab和Li2Sx的能量。
2 结果与讨论
2.1 吸附构型与吸附能
已报道的研究中,石墨烯对于多硫锂化合物的吸附能力较差,仅为-0.31 eV~-0.57 eV,其中对Li2S的吸附能的结合能最高(-0.57 eV)。 较低的吸附能数据表明,石墨烯作为电极材料并不能有效吸附锂硫化合物。
用DFT计算催化剂与硫的结合能。Fe3O4能与Li2S4和Li2S2等多硫化物形成较强的吸附,远大于对于Li2S6和Li2S的吸附。进一步研究了上述多种锂硫化合物在N/Fe3O4表面的吸附模型,探讨了N掺杂Fe3O4与硫锂化合物结合强度的变化。
研究了Li2Sx(x=1、2、4和6)在Fe3O4和N/Fe3O4上的吸附构型,稳定的吸附结构如图1所示。计算了吸附能数据,如图2所示。

图1 Li2Sx在N/Fe3O4上的吸附稳定构型Fig.1 Stable adsorption structure of Li2Sx on N/Fe3O4

图2 Li2Sx分别在Fe3O4和N/Fe3O4上的吸附能Fig.2 Adsorption energy for Li2Sx on Fe3O4 and N/Fe3O4
Li2Sx(x=1、2、4和6)在Fe3O4上吸附时,吸附能处于-1.325 eV~-2.874 eV。在N/ Fe3O4上进行吸附时,计算获得的吸附能从-2.28 eV~-3.88 eV。进一步加强了对锂硫化合物的吸附作用,吸附数据表明,N/Fe3O4可能是一种理想的电极材料。
结构优化后,分析Fe3O4和N/Fe3O4吸附的锂硫化合物中涉及化学作用的键长数据。很明显可以看到,在N掺杂前后,距离锂硫化合物最近的Fe-O键变短,由此可以推断出在N加入体系后,加强了Fe-O的键和作用,增强了电极本身的吸附能力。研究发现,N/Fe3O4在吸附Li2S6和Li2S4后,电极材料Fe3O4和多硫化合物之间形成的Fe-S键变短。具体来说,硫与N/Fe3O4和Fe3O4形成Fe-S键。此外,Fe-S在N/Fe3O4-可溶性多硫化物(Li2S6和Li2S4)中的键长及N/Fe3O4-Li2S2/N/Fe3O4-Li2S放电产物中的键长均短于原始Fe3O4,有利于促进多硫锂化合物的催化反应,抑制了多硫化物的穿梭溶解行为,减缓了Li2S的分解过程,这一系列变化提高了整体的催化转换效率。
为了更深层次地分析实现N掺杂前后对于电极材料吸附的本质原因。着重分析了放电过程中的Li2S4和充电过程中的Li2S的键长变化情况。在N/Fe3O4吸附Li2S4中,S-S键的长度为2.172 Å,而Fe3O4吸附Li2S4中S-S键的长度为2.117 Å。相对应的,Li2S键中Li-S键从Fe3O4中的2.364 Å加长到了N/Fe3O4的2.541 Å。以上说明N掺杂可以、更加有效削弱S-S键和Li-S键的强度。同时,在N/Fe3O4中,由于N和O原子的电负性不同,且化学作用力新增Li-N离子键。与Fe3O4相比,N/ Fe3O4对锂硫化合物表现出更大的吸附能,这是由于N的缺电子性质导致N/ Fe3O4具有更强的极化作用。
结合吸附能数据和键长变化结果可知,推断出多硫锂化合物硫Fe3O4和N/Fe3O4表面可能的转化机制。在放电过程中,Li2S6分子吸附在N/Fe3O4表面,同时与N/Fe3O4形成强的Fe-S键,随后继续发生反应,逐渐在N/Fe3O4表面转化为Li2S4。Li2S4与N/Fe3O4催化剂形成较强的Fe-S键,S-S桥键被拉长和减弱。随着更多的S-S键的断裂和多硫锂化合物的转换,形成了最终产物Li2S。在充电过程中,Li2S与N/Fe3O4形成较强的Fe-S键,Li-S键容易被拉伸和减弱,分解为LiS和Li,最后逐渐转化为硫。
2.2 总态密度
为了研究掺杂前后Fe3O4对于电子态密度的影响,对态密度进行了分析。研究掺杂前后对于态密度的相对影响程度,集中关注成键状态和费米能级处的情况。如图3,通过比较发现,对于Fe3O4和N/Fe3O4,掺杂N后,带隙的宽度明显降低,两者在费米能级处均处于电子态密度中,属于金属性质。可以认为是N的加入使得更多电子由非占据态转换为占据态,促使费米能级向着更高能级移动,出现了新的电子态密度,在降低带隙的同时,增加了体系自身的导电性。
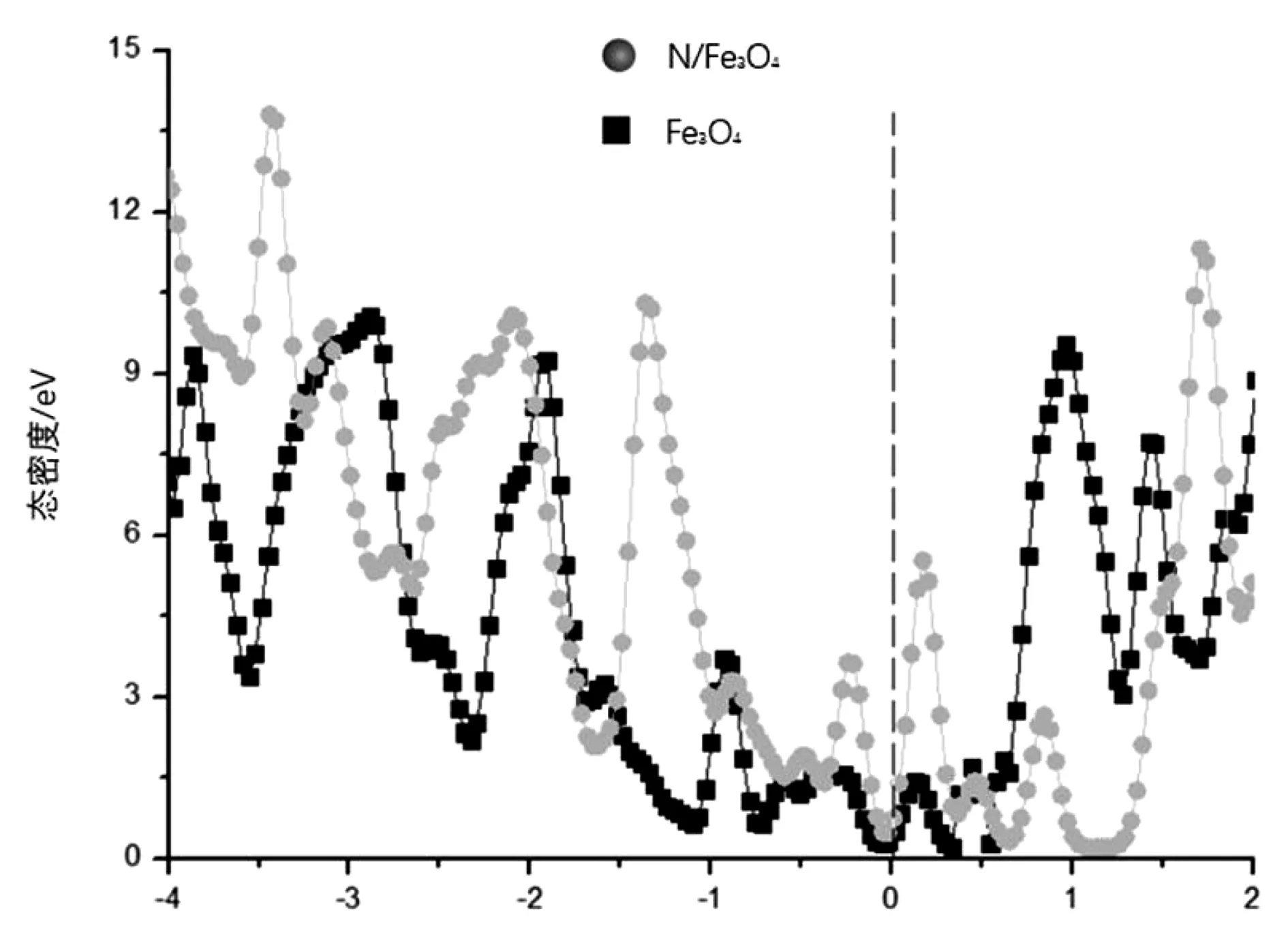
图3 Fe3O4和N/Fe3O4的总态密度Fig.3 State density of Fe3O4 and N/Fe3O4
掺杂N之后,与周围的Fe之间互相成键,由此而产生新的态密度。新的电子态密度的产生降低了带隙,使得电子发生跃迁所需的能量降低,有利于电子转移,提高了掺杂体系的导电率。
N/Fe3O4中总态密度波段中心明显向更高的能量转移,与Fe3O4相比,明显更接近费米能级,说明N/Fe3O4催化剂在硫催化过程中具有更高的载流子密度和良好的电荷转移。同时,总态密度的中心越靠近费米能级下,反键态填充越少,对多硫化物的化学吸附越强。
2.3 离子扩散动力学
Li+在正极材料中的脱嵌过程直接影响电池本身的放电倍率性能。在Li+扩散路径中,由于电荷作用会发生极化现象,将影响离子的转移过程。
为此,模拟Li+转移过程,计算在扩散过程中所发生的能量变化过程。计算了Li+在Fe3O4和N/Fe3O4上的扩散能垒,结果如图4所示。Li+在N/Fe3O4上的扩散能垒仅为0.33 eV,远小于在Fe3O4(0.71 eV)。

图4 在Fe3O4和N/ Fe3O4上,Li+的扩散能垒Fig.4 Energy barrier of Li+ on Fe3O4 and N/ Fe3O4
结果显示,N掺杂可以有效降低扩散能垒,并且新生成的Fe-N键促进了Li+的扩散。
通过操纵N掺杂成功构建了一种新型的N/Fe3O4双功能催化剂。N/Fe3O4能同时促进放电过程中的多硫化物转化反应和充电过程中的Li2S氧化。
理论模拟分析结果表明,N的掺杂改变了催化剂的表面电子结构。DFT计算结果还揭示了硫在双官能团表面的可能转化机制。这表明设计和构建N掺杂的双功能催化剂可以提高锂硫电池电池的电化学性能。通过了解离子掺杂对催化剂电子结构和配位环境的调节,将为设计新型催化剂实现高性能锂硫电池提供新的思路。
3 结论
采用密度泛函理论对锂硫电池正极材料Fe3O4和N/Fe3O4对多硫化合物的吸附和催化活性进行理论研究。
从能量角度分析,经过掺杂后,Fe3O4和不饱和N原子之间强烈化学键作用使得N原子周围成为催化活性中心,致使N/Fe3O4表现出更强的金属特性,出现了新的杂质能级,可以起到降低能带带隙和提高导电性能的作用。
从键合作用分析来看,对N/Fe3O4体系而言,Fe-O键和Fe-S键变短,增强了对于多硫化合物的吸附。而对于多硫化合物的催化过程,S-S键变长,促进了多硫化合物之间的相互转换。此外,N的掺杂可以有效降低Fe3O4扩散能垒,提高电池本身的倍率性能。
猜你喜欢
杭州(2023年3期)2023-04-03 07:22:04
装备制造技术(2020年4期)2020-12-25 05:25:52
复旦学报(医学版)(2020年3期)2020-06-18 07:36:52
原子与分子物理学报(2020年5期)2020-03-17 07:00:00
中国资源综合利用(2016年7期)2016-02-03 03:00:11
环境科技(2015年3期)2015-11-08 12:08:36
电源技术(2015年2期)2015-08-22 11:28:30
电源技术(2015年5期)2015-08-22 11:17:54
电源技术(2015年9期)2015-06-05 09:36:06
应用化工(2014年11期)2014-08-16 15:59:13
